 |
BMe Research Grant |

|

George A. Olah Doctoral School in Chemistry and Chemical Technology
Department of Inorganic and Analytical Chemistry
Supervisor: Dénes Szieberth PhD.
Hydrogen activation with non-metals
Introducing the research in a few words
In industrial and laboratory practice it is of common occurrence that we would like to saturate molecules having unsaturated double and triple bonds with hydrogen. The cleavage of the H−H bond is very hard so the activation of the hydrogen (increase of the reactivity) is very important in different fields of chemistry: petrochemistry, pharmacochemistry food industry, hydrogen storage[1]. Usually, transition metals are used as a catalyst for the cleavage of hydrogen. Nowadays the usage of the non-metal catalysis is developing extremely fast because of the possible high price and the environmental hazard of the transition metals. During my PhD research such non-metallic hydrogen catalysts have been studied using different computational chemistry techniques.
Brief introduction of the research group
This research is carried out by the group of Prof. László Nyulászi at the BUTE, Faculty of Chemical Technology and Biotechnology, Department of Inorganic and Analytical Chemistry. The main interests of the theoretical chemistry group are ionic liquids, carbene chemistry, main group elements chemistry, organocatalysis. Results are regularly published in leading scientific journals.

Theoretical chemistry group (BUTE)
Broader presentation of the history of the research
Nowadays hydrogen activation with non-metal elements is an intensively researched field of chemistry[2,3,4]. For the design of such organocatalysts the usage of the Frustrated Lewis Pair (FLP) theory is preferred[5]. The FLPs have a Lewis acid and a Lewis base functional group, which cannot make a dative bond with each other because of the bulky substituents attached. The resulting reactive center in the molecule pair can easily activate small molecules, for example the dihydrogen (Fig. 1). Furthermore, in some cases these activation reactions are reversible[5,6]. The reversibility is very important because the catalysts should be able to release the hydrogen, thus the excessive stability of the products is undesirable.
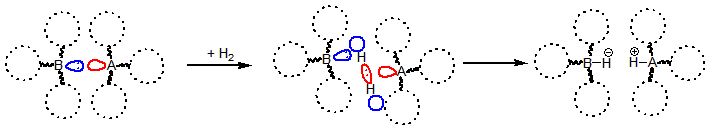
Figure 1: Hydrogen activation with Frustrated Lewis Pairs
The mechanism of the hydrogen activation with FLPs differs from the transition metals (Fig. 2). In the case of metals the reaction follows a stepwise mechanism and an intermediate appears on the reaction pathway[7].
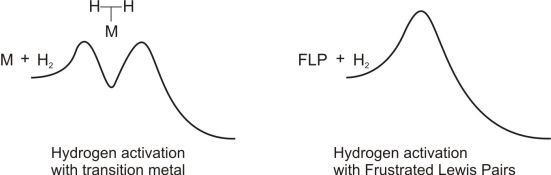
Figure 2: Two possible mechanisms of the hydrogen activation (left side: with transition metal, right side: with FLP)
The stabilization of the η2-H2 complex intermediate arises from the superposition of two interactions: the donation of the σ-bond of the dihydrogen to an empty orbital of the metal, and the backdonation of a d-type orbital of the metal to the σ*-orbital of the dihydrogen[7]. The elongation of the H−H bond – thus the activation of dihydrogen – is the consequence of these two bonding interactions.
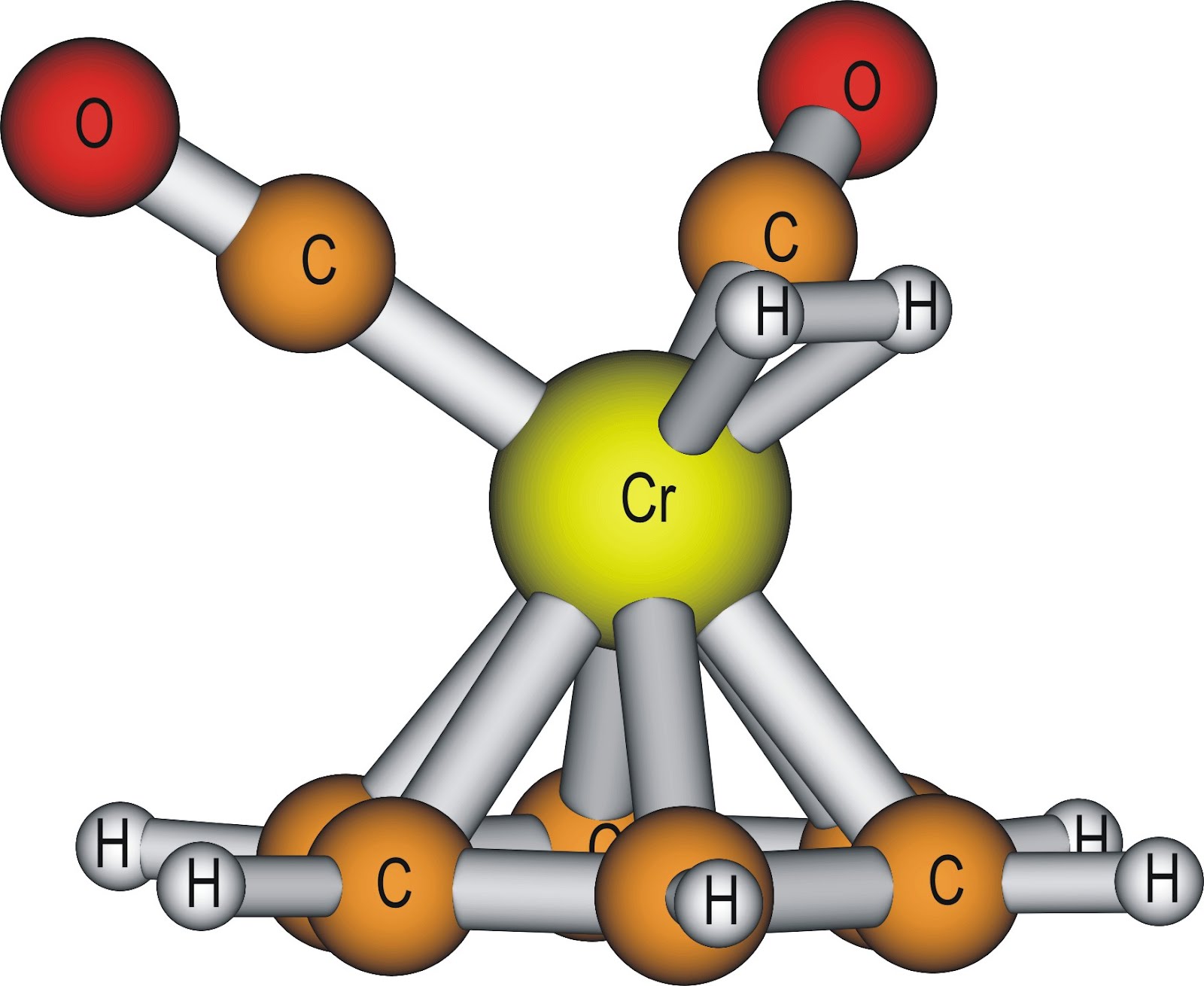
Figure 3: (η6-benzene)(CO)2Cr(η2-H2) complex is an example of an η2-H2 complex[8]
The goal of the research, and possible issues
In the case of FLPs the hydrogen activation reaction follows a one-step mechanism where no η2-H2 complex is present on the reaction pathway[9]. Contrary to the transition metals, in the case of FLPs the Lewis acid (electron acceptor) and the Lewis base (electron donor) are not localized on one center. The strong polarization effect of these two centers (Lewis acid and Lewis base) results in the cleavage of the H−H bond, and the η2 complex generally cannot be formed[9]. In our previous study we investigated a unique diphosphine-borane system where – contrary to the expectations – an η2-H2 complex forms during a hydrogen activation (see later: Previous results). Thus a new type of reaction route takes place: a non-metallic element can imitate the behavior of transition metals (Fig. 4).
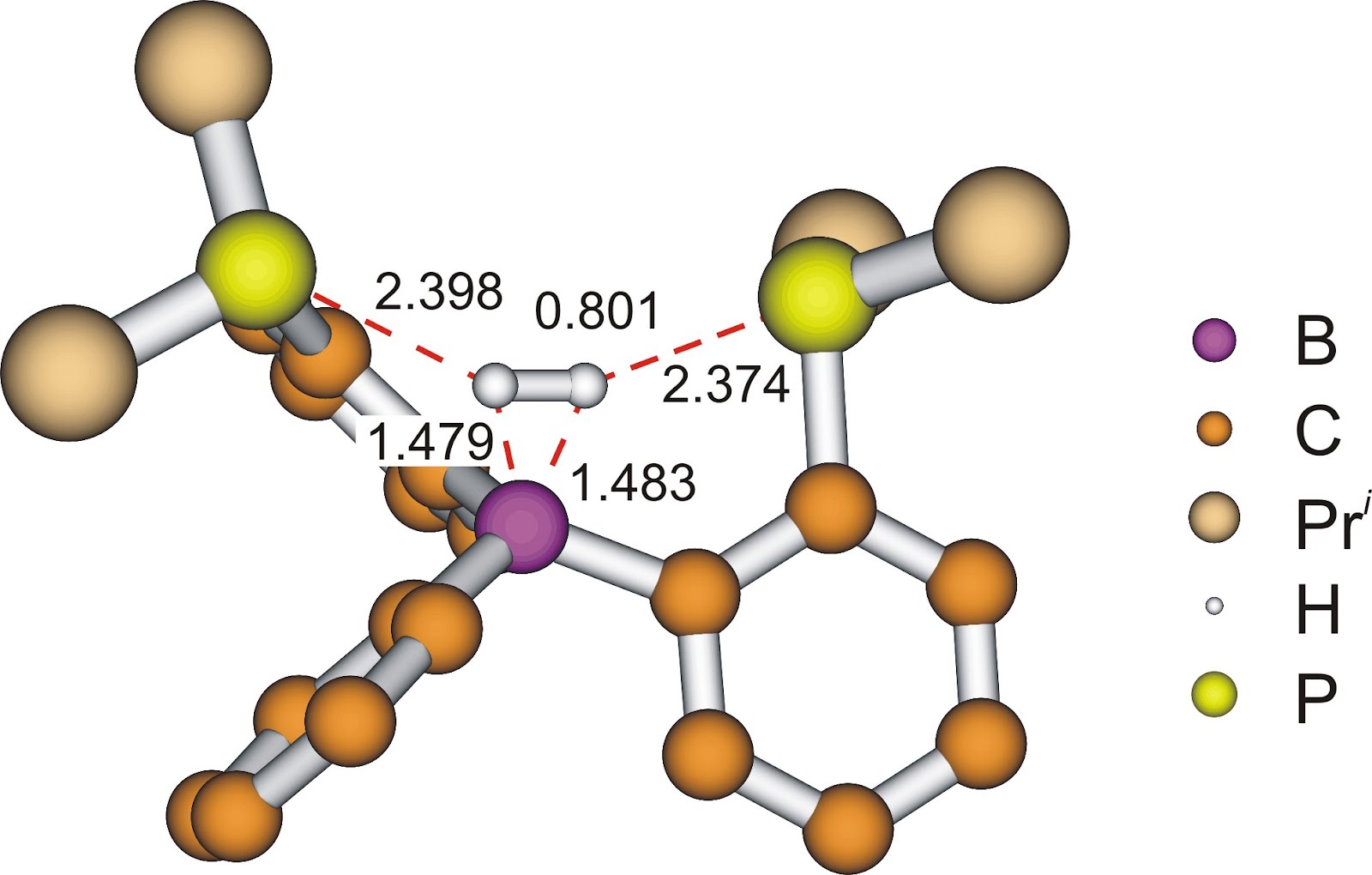
Figure 4: η2-H2 complex of the diphosphine-boranes
One of the goals of the planned research is to investigate the effect of different substituents on the Lewis centers on the stability of the η2-H2 diphosphine-borane and the reaction barrier. Our aim is to find the substituent combination that gives the lowest activation barrier while preserving reversibility. In parallel we would like to study how we can stabilize the η2-H2 BR3 complex without the help of external electron donor groups. In the literature there are only a few examples of η2-H2 borane complex so far, furthermore only BH5 can be prepared[10].
Methods
To answer the above mentioned questions we would like to use different ab initio quantum chemistry methods. With the help of the theoretical chemistry methods the structure of the molecules and the mechanisms of reactions can be easily explored. The stability of a complex can be described in several ways. For example, we can analyze the bond lengths in the molecule or through the analysis of the electron density the bond order can be calculated (AIM). We can also calculate the energy difference between the complex and its separated components or with the Natural Bond Orbital analysis (NBO) we can estimate the interaction between two molecule fragments. For a complete description of the stability of the η2-H2 complex we would like to use all of these methods.
Before using a quantum chemical model, the eligibility of the model must be checked. Therefore, precalculations must be made to choose the proper level of theory (basis set and method) which describes well the problem. Because of the large size of the system the Density Functional Theory (DFT) seems to be a good compromise between accuracy and calculation time (better result → longer calculation time). From several available DFT methods the selection of the proper one will be carried out by comparison with higher level ab initio test calculations (MP2, CCSD(T)) on model molecules.
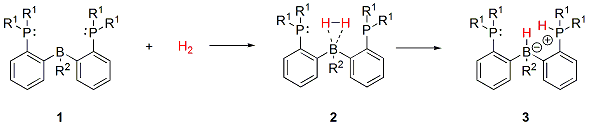
Figure 5: The reversible hydrogen activation of diphosphine-borane
R1: −CH3, −iPr, −tBu, −SiH3, −NiPr2 ; R2: −H, −CH3, −Ph, −Cl, −CF3, −SiH3, −SiMe3
Our previous study suggests that the electron donor character of the phosphorus atoms and the electron acceptor character of the boron atom have to be strengthened for the stabilization of the diphosphine-borane[11]. Therefore we would like to calculate the reactions shown on Figure 5 by several substituents having different steric and electronic effects: R1: −CH3, −iPr, −tBu, −SiH3, −NiPr2 ; R2: −H, −CH3, −Ph, −Cl, −CF3, −SiH3, −SiMe3. Furthermore, we want to examine the effects of the replacement of the Lewis acid and base centers (eg.: boron to aluminum, phosphoruses to nitrogens).
We would like to calculate the following reaction (BR3 + H2 = BR3H2) with several R substituents having different electronic and steric effects for the study of the stability of the η2-H2 BR3 complex. The BR3H2 η2-H2 borane complexes are very unstable and decompose easily. The instability of these complexes can be explained by the lack of the d-type lone pair of the boron, thus the boron cannot (back)donate electron to the σ*-orbital of the H2 molecule. The stability of the molecule can be described by calculating the energy of the above mentioned reaction.
Previous results
In our previous work we showed that the diphosphine-borane complexes could activate dihydrogen reversibly[11] (Fig. 5). The diphosphine-borane is an FLP-type molecule which has one Lewis acid (electron acceptor) and two Lewis base (electron donor) functional groups[12]. Because of the symmetrical arrangement of these functional groups the diphosphine-borane can imitate the η2-H2 complex of the transition metals (Fig. 4 or molecule 2 on Fig. 5). Theoretical chemistry computations show that there are two main interactions in the complex: first the two lone pairs of the phosphorus atoms symmetrically donate electron to the hydrogen molecule, secondly the resulting excess electrons are donated to the empty orbital of the boron. The combined effects of these electron transfers strengthens the B−H2 bond[11]. This electron transfer can be traced by the topological analysis of the electron density of the complex. 3D maps can be made from the electron density where the chemical bonds are assigned to the maxima of the density and the bond critical points. On Figure 6 the η2-H2 complex of the diphosphine-borane is shown. The bond paths between the two phosphorus atoms and the hydrogens and between the dihydrogen and the boron are shown with dotted line. The calculations also show that the stability of the intermedier significantly affects the barrier height of rate-determining step.
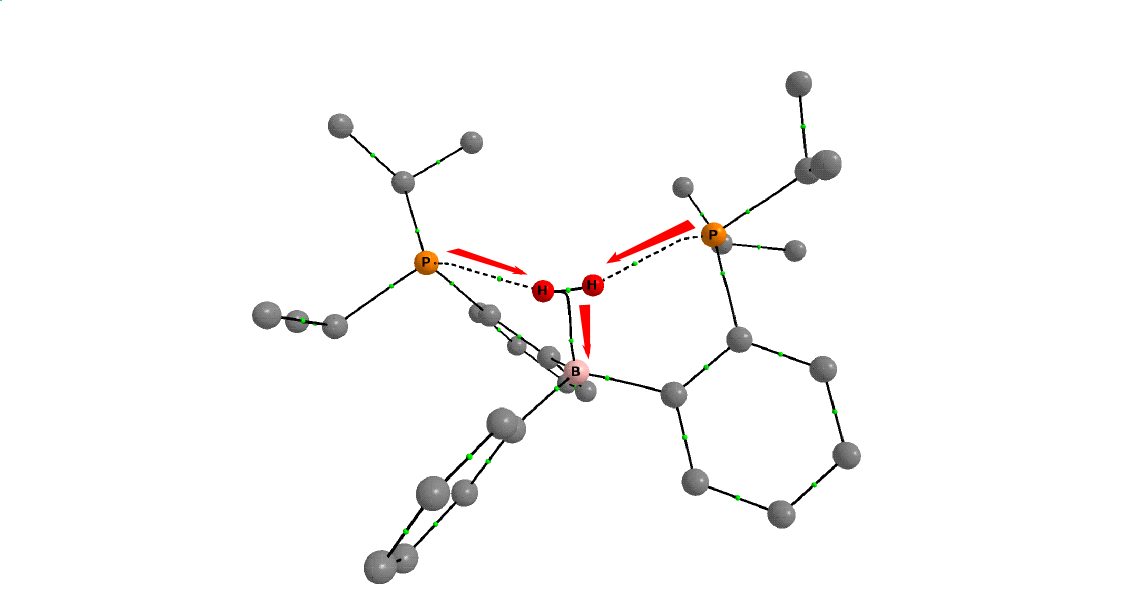
Figure 6: AIM plot of the η2-H2 diphosphine-borane complex
Expected impact and further research
According to the computational results we would like to show a new non-metallic hydrogen activation mechanism which can introduce a new way of thinking about the FLP chemistry. The second electron donor group in the molecule can prompt the development of new type of non-metallic catalysts which can further reduce the barrier of hydrogen activation. We would like to synthesize the possible target compounds in a cooperation with a research group in Toulouse. Furthermore, according to the computations we can hope to predict stable η2-H2 borane complexes – a very interesting outcome knowing that until now there are only a few examples in the literature for pentavalent boron atom.
Own publications, references, links
Publications:
L. Könczöl, A. Kawachi, D. Szieberth, Organometallics 2012, 31, 120-125
L. Könczöl, E. Makkos, D. Bourissou, D.Szieberth, Angew.Chem Int. Ed. submitted
Links:.
References:
[1] J. F. Hartwig, in Organotransition Metal Chemistry: From Bonding to Catalysis; University Science Books: Sausalito, CA 2010
[2] a) A. L. Kenward, W. E. Piers, Angew. Chem. 2008, 120, 38–42; Angew. Chem. Int. Ed. 2008, 47, 38–41. b) D. W. Stephan, Chem. Commun. 2010, 46, 8526–8533. c) P. P. Power, Nature 2010, 463, 171–177. d) P. P. Power, Acc. Chem. Res. 2011, 44, 627–637
[3] a) G. H. Spikes, J. C. Fettinger, P. P. Power, J. Am. Chem. Soc. 2005, 127, 12232–12233. b) Y. Peng, M. Brynda, B. D. Ellis, J. C. Fettinger, E. Rivard, P. P. Power Chem. Commun. 2008, 6042–6044. c) Y. Peng, B. D. Ellis, X. Wang, P. P. Power, J. Am. Chem. Soc. 2008, 130, 12268–12269. d) Y. Peng, J.-D. Guo, B. D. Ellis, Z. Zhu, J. C. Fettinger, S. Nagase, P. P. Power, J. Am. Chem. Soc. 2009, 131, 16272–16282. e) Z. Zhu, X. Wang, Y. Peng, H. Lei, J. C. Fettinger, E. Rivard, P. P. Power, Angew. Chem. 2009, 121, 2065–2068; Angew. Chem. Int. Ed. 2009, 48, 2031–2034
[4] a) G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller, G. Bertrand, Science 2007, 316, 439–441. b) D. Martin, M. Soleilhavoup, G. Bertrand, Chem. Sci. 2011, 2, 389–399
[5] a) G. C. Welch, R. R. San Juan, J. D. Masuda, D. W. Stephan, Science 2006, 314, 1124–1126. b) P. A. Chase, G. C. Welch, T. Jurca, D. W. Stephan, Angew. Chem. 2007, 119, 8196–8199; Angew. Chem. Int. Ed. 2007, 46, 8050–8053. c) G. C. Welch D. W. Stephan, J. Am. Chem. Soc. 2007, 129, 1880–1881. d) P. Spies, G. Erker, G. Kehr, K. Bergander, R. Fröhlich, S. Grimme, D. W. Stephan, Chem. Commun. 2007, 5072–5074. e) P. Spies, S. Schwendemann, S. Lange, G. Kehr, R. Fröhlich, G. Erker, Angew. Chem. 2008, 120, 7654–7657; Angew. Chem. Int. Ed. 2008, 47, 7543–7546
[6] D. W. Stephan, G. Erker, Angew. Chem. 2010, 122, 50–81; Angew. Chem. Int. Ed. 2010, 49, 46–76
[7] a) R. H. Crabtree, Angew. Chem. 1993, 105, 828–845; Angew. Chem. Int. Ed. 1993, 32, 789–805. b) G. J. Kubas, in Metal dihydrogen and sigma–bond complexes; Kluwer Academic/Plenum ed.; New York, 2001. c) S. Aldridge, A. J. Downs, Chem. Rev. 2001, 101, 3305–3365. d) G. J. Kubas, Chem. Rev. 2007, 107, 4152–4205. e) R. D. Adams, B. Captain, Angew. Chem. 2008, 120, 258–263; Angew. Chem. Int. Ed. 2008, 47, 252–257. f) G. J. Kubas, J. Organomet. Chem. 2009, 694, 2648–2653
[8] J. D. Egbert, D. M. Heinekey, Organometallics 2010, 29, 3387–3391
[9] a) T. A. Rokob, A. Hamza, A. Stirling, T. Soós, I. Papai, Angew. Chem. 2008, 120, 2469–2472; Angew. Chem. Int. Ed. 2008, 47, 2435–2438. b) R. Rajeev, R. B. Sunoj, Chem. Eur. J. 2009, 15, 12846–12855. c) S. Grimme, H. Kruse, L. Goerigk, G. Erker, Angew. Chem. 2010, 122, 1444–1447; Angew. Chem. Int. Ed. 2010, 49, 1402–1405
[10] a) T. J. Tague Jr., L. Andrews, J. Am. Chem. Soc. 1994, 116, 4970–4976. b) P. R. Schreiner, H. F. Schaefer III, P. v. R. Schleyer, J. Chem. Phys. 1994, 101, 7625–7632. c) J. D. Watts, R. J. Bartlett, J. Am. Chem. Soc. 1995, 117, 825–826. d) S. Fau, G. Frenking, Mol. Phys. 1999, 96, 519–527. e) O. A. Filippov, A. M. Filin, V. N. Tsupreva, N. V. Belkova, A. Lledós, G. Ujaque, L. M. Epstein, E. S. Shubina, Inorg. Chem. 2006, 45, 3086–3096. f) C. Fan, L. G. Mercier, W. E. Piers, H. M. Tuononen, M. Parvez, J. Am. Chem. Soc. 2010, 132, 9604–9606. g) Z. Lu, Z. Cheng, Z. Chen, L. Weng, Z. H. Li, H. Wang, Angew. Chem. 2011, 123, 12435–12439; Angew. Chem. Int. Ed. 2011, 50, 12227–12231. h) G. I. Nikonov, S. F. Vyboishchikov, O. G. Shirobokov, J. Am. Chem. Soc. 2012, 134, 5488–5491. d) J.-L. M. Abboud, B. Németh, J.-C. Guillemin, P.Burk, A. Adamson, E. R. Nerut, Chem. Eur. J. 2012, 18, 3981–3991
[11] L. Könczöl, E. Makkos, D. Bourissou, D.Szieberth, Angew.Chem Int. Ed. submitted
[12] S. Bontemps, G. Bouhadir, P. W. Dyer, K. Miqueu, D. Bourissou, Inorg. Chem. 2007, 46, 5149–5151