 |
BMe Research Grant |

|

BME, György Oláh Doctoral School
Richter Gedeon Plc.
Supervisor: Dr. György M. Keserű
Computational Models for Drug Discovery
Introducing the research area
The most desired objective of drug discovery is delivering efficient and safe drugs to exert significant contribution to human welfare. Due to the immense complexity of the underlying biochemical processes of the human body, medicinal chemistry faces a multidimensional optimization task. During my studies, I was working on computational methodologies and concepts to aid the rational and efficient design of novel compounds.
Brief introduction to the research site
Gedeon Richter Plc. has one of the largest original drug research facility in Middle-Europe. The quality of the research that was made during the last decades is highlighted both in well-known original drugs such as Cavinton and Tolperisone and also through the recent example of Cariprazine that is filed for administration after positive human phase III clinical trials both in schizophrenia and mania.
History and context of the research
Compounds in the preclinical research phase are assessed by the set of numerous measured in vitro parameters. During the clinical trials, efficacy is influenced by the interactions with the target (affinity, functional response, binding kinetics) and also by the pharmacokinetic parameters, see Figure 1. [1-2]. Drug safety crucially depends on the modification of side-effect related biochemical processes such as inhibition of physiologically important enzymes and receptors. Out of the more than 30 000 genes [3] encoded in the human genome that express proteins able to bind drug-like molecules, drugs are to interact with one or a few representatives. The possible side effects incorporate the blockade of hERG ion channel causing arrhythmia, inhibition of cytochrome enzyme system leading to hepatotoxicity or the interaction with cell membranes that results in cellular toxicity (phospholipidosis) [4].
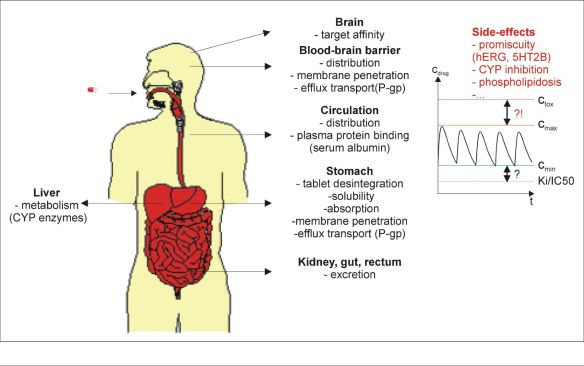
Figure 1. Fate of drugs in human body.
Solving the Rubik’s cube is a witty analogy for the optimization task[5]. The cube has 54 outer squares in six different colors, allowing ~43*1018 different configurations (still dwarfed by the astronomic size of the drug-like chemical space (1060)), but there is no initial configuration of the cube that cannot be solved in maximum 20 moves. During the course of medicinal chemistry optimizations typically 100-3000 compounds are synthesized, although it can be proved by retrospective analysis that final compound can be achieved in less than 20-60 steps, underlining the importance of finding and efficient strategy (see Figure 2. )[6].
Drug design involves computational approaches to find general rules (remarkable Lipinski-rules[7]) and develop methodologies to predict the characteristics (affinity, toxicity) of the molecules ahead of the synthesis.
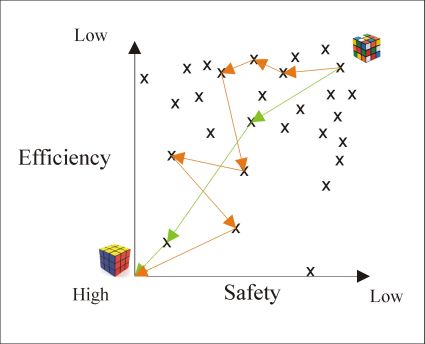
Figure 2. Schematic view of medicinal chemistry optimization. Optimization on the green path is more effective and rational, than the orange trajectory.
The research goal, open questions
During my research, questions were conceived from two different aspects: on atomic level and from a more holistic perspective. First, two proteins with significant involvement in the pharmacokinetic behavior of drugs, the cytochrome P450 (CYP) enzyme family and the P-glycoprotein were selected for atomic level simulations.
Breakthroughs in protein engineering and x-ray crystallography during the last decade have yielded several protein structures with outstanding pharmaceutical relevance. First structure of human CYP family to publish was the CYP2C9 [8] in 2003, and in 2009 the structure of mouse P-glycoprotein (P-gp) [9] was resolved which opened the door to using structure-based approaches.
-
My aim was to develop and test a novel, structure-based site of metabolism prediction methodology.
-
Publication of the mouse P-gp structure prompted me to build and validate the homology model of the human orthologue.
-
From a more broader perspective, my plan was to assess the compound quality in terms of the relationships between affinity and physicochemical parameters to find the optimal values.
Methods
Due to the large number of protein heavy atoms, they can be best studied computationally, typically using molecular mechanics.
Docking is a conformational search of small molecules in the binding site of proteins, that determines the binding mode and the corresponding binding energy. During my studies, I used Glide fast-docking tool, a validated and high performance tool widely used as part of the computational drug design software arsenal[10]. Docking typically considers ligand flexibility but handles proteins atoms rigidly. Induced Fit Docking (IFD) is a special case of docking, where binding site flexibility is incorporated at the expense of longer computational time.
The aim of homology modelling is to predict a protein structure from its sequence by using a structure of a different protein with high sequence similarity to the target. This prediction is based on the parallel observation that similar sequences adopts approximately identical structures.
Lipophilicity is one of the crucial parameters used in drugs discovery. Measured as the logarithm of the octanol−water partition coefficient (logP) or as the pH-dependent distribution coefficient (logD), it contributes to pharmacokinetics and toxicological risk factors[11-12]. Lipophilic ligand efficiency metrics such as LLE and LELP congregates potency and lipophilicity in a single index, to address both properties simultaneously. Leeson and Springthorpe introduced LLE[13], defined as the difference of log P (or log D) and the negative logarithm of a potency measure (pKi, or pIC50/pEC50). The concept of lipophilicity-corrected ligand efficiency, LELP, was first introduced by Keserű and Makara [14]. It is defined as the ratio of logP and ligand efficiency (LE=∆Gbind/(Heavy atom count)). Therefore, higher LLE and lower LELP values indicates more desirable lipophilic efficiency.
Results
Site of metabolism prediction
X-ray crystallographic studies of the catalytic cycle of CYP 450cam revealed that the substrate (camphor) has limited mobility during the reaction [15] (Figure 3.). This exciting observation has led to the proposed product- or reverse-docking approach. Since the key point is overlap, the atom introduced during biotransformation must be in the vicinity of the catalytic site. Comparing to the traditional, substrate orientation-based methods, the extracted spatial constraint provides extra information, that can be exploited to enhance the conformational search (Figure 4.)[T1].
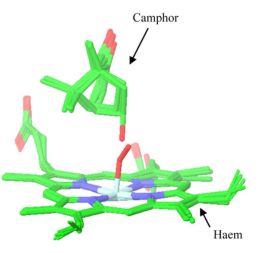
Figure 3. Substrate, intermediate and product structures of P450cam camphor hydroxylation [15].
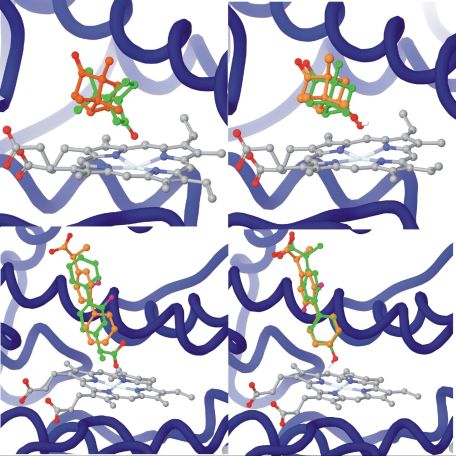
Figure 4. Comparison of substrate docking and product docking[T1]. The first and second row shows the docking for camphor (P450cam) and flurbiprofen (CYP2C9) cases, respectively. The left column is the substrate and to the right is the product docking (orange) compared to the x-ray structure (green). In both cases, product docking has higher alignment to the experimental binding mode.
The reverse-docking approach involves three steps: (i) generation of possible metabolites for a query compound, (ii) docking the possible metabolites into the binding site of the enzyme, and (iii) selection of the most probable metabolites based on two orientations and complementarity to the binding site.
As a proof of concept, I evaluated the methodology on human CYP2C9 isoform. Docking was able to identify the correct metabolite among the three best-ranked compounds in 83.7% of the cases, outperforming the substrate docking results (47%). The predictive power of the developed methodology was comparable to the approaches based on alternative theories as described in literature [T2]. Advantage of the reverse-docking method is to result an exact metabolite structure instead of merely predicting the site of metabolism, and adaptation does not require dedicated software.
The phenomenon of cooperative binding is well-known in the case of CYP enzymes (see CYP3A4 complexed with ketoconazole). Our group developed a sequential docking methodology to predict the binding mode of cooperative binders[T3] and might contribute to atomic level understanding of this unique behaviour.
Homology modelling of P-gp
Human P-gp structure was predicted based on the mouse orthologue by homology modelling [T4]. The obtained model was validated in terms of stereo-chemical (Ramachandran-plot) and energetic (ProSa) parameters. Four of the biochemically most studied ligands were docked to the binding site using IFD. The binding modes thus obtained were in line with the experimental results. In contrast, virtual screening using rigid protein assumption yielded poor results. Based on these studies, it was concluded that incorporation of protein flexibility is highly important in the case of P-gp, therefore rigid docking into the homology model is not suitable for large-scale virtual screening purposes.
Promiscuity
Side effects of drugs are critically influenced by the affinity profile to non-target proteins (promiscuity). Summary of the literature data highlighted that lipophilicity and basic character are the most important physicochemical parameters correlating with promiscuity (Figure 5.) [T5]. Less lipophilic compounds (calculated logP<4) were found to have significantly lower hit rates compared to more lipophilic (calculated log>4) ones (Student t-test, p<0.001) on my particular dataset.
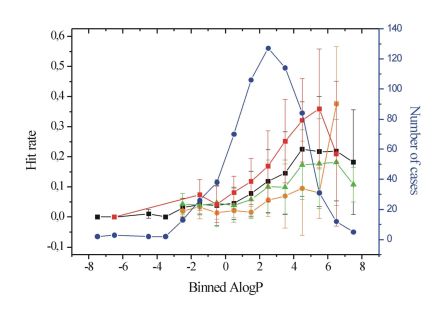
Figure 5. Plot of lipophilicity (AlogP) versus hit rate[T5].
The collected knowledge suggests rigorous use of the predicted physicochemical parameters and implicates a design work-flow similar to that depicted in Figure 6. [T5]. Given the crucial role of acid-base properties and ionization constant, I have evaluated the accuracy of in silico pKa prediction tools[T6].
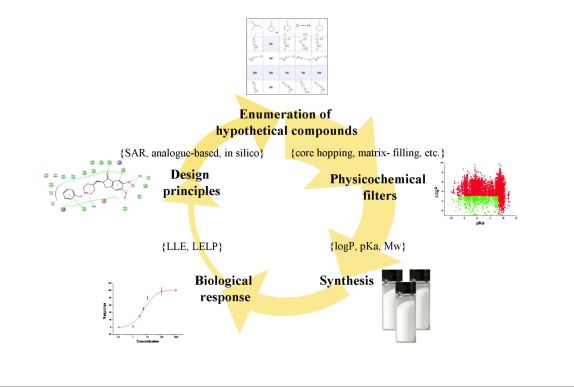
Figure 6. Proposed medicinal chemistry optimization cycle[T5].
Lipophilic efficiency
I investigated the distribution of LLE and LELP values on multiple data sets representing different stages of preclinical drug discovery (Figure 7. )[T7].
The results underlined the importance of using LLE and LELP metrics, since the compounds having successful clinical phase I. results, and the marketed drugs could be separated from the preclinical compounds. Therefore, a desirable preclinical optimization strategy would include the monitoring of lipophilic efficiency and the use of these values as main driving forces during design to achieve the domain of successful examples.
Linking lipophilic efficiency and pharmacokinetic assay results revealed that LELP had higher correlation with the propensity of violating assay criteria compared to LLE. Investigation of the effects of various substituents resulted in an operation scheme for LELP-based optimizations that might be useful in medicinal chemistry practice.
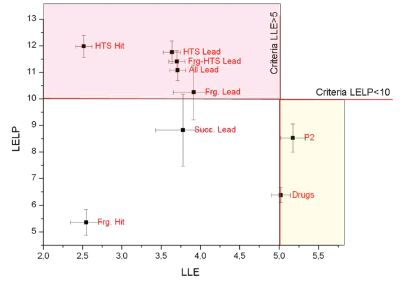
Figure 7. LELP versus LLE for the different subsets of drug research. All data are mean values with error bars representing the standard error values[T8]. Hit: hit compounds, Lead: lead compounds, Frg: fragment optimization compounds, HTS: high-throughput screening compounds , P2: human phase II. compounds, Drugs: marketed drugs.
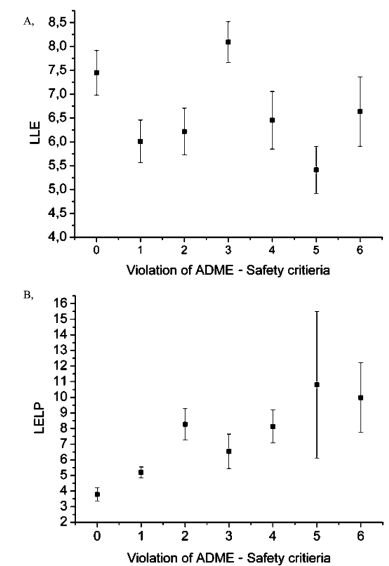
Figure 8. Mean LLE (A) and LELP (B) values for compounds violating a given number of in vitro ADME or safety end-point criteria . Error bars represent the standard error values[T7].
Expected impact and further research
According to the feedbacks, the scientific community received my work with great interest. Two papers, [T5] and [T7], were listed among the 20 Most Read Articles of Journal of Medicinal Chemistry considering both the 1 month and 1 year statistics, and were among the 6 most read articles of the first quarter of the year. The paper dealing with lipophilic efficiency [T7] was evaluated by John Lowe and recommended in the F1000Prime selection[16].
My papers that are incorporated in this study had been cited more than 60 times to date.
Henceforth, I plan to study the relationship between binding thermodynamics and selectivity, based on the hypothesis of Freire and co-workers[17].
Publications, references, links
Publications
[T1] Tarcsay, A.; Kiss, R.; Keserű, G.M., Site of metabolism prediction on cytochrome P450 2C9: a knowledge-based docking approach. J Comput Aided Mol Des 2010, 24,pp. 399-408. (IF:3.386)
[T2] Tarcsay, Á.; Keserű, G.M., In silico site of metabolism prediction of cytochrome P450-mediated biotransformations. Expert Opin Drug Metab Toxicol. 2011, 7, pp. 299-312. (IF:3.119)
[T3] Vass, M.; Tarcsay, Á.; Keserű, G.M., Multiple ligand docking by Glide: implications for virtual second-site screening. J Comput Aided Mol Des. 2012, 26, pp. 821-34. (IF:3.386)
[T4] Tarcsay, A.; Keserű, G.M., Homology modelling and binding site assessment of the human P-glycoprotein. Future Med Chem. 2011, 3,pp. 297-307. (IF:2.522)
[T5] Tarcsay, Á.; Keserű G.M., Contributions of molecular properties to drug promiscuity.
J Med Chem. 2013, 56, pp. 1789-1795. (IF: 5.248)
[T6] Balogh, G.T.; Tarcsay, A.; Keserű, G.M., Comparative evaluation of pK(a) prediction tools on a drug discovery dataset. J Pharm Biomed Anal. 2012 pp. 67-68, 63-70. (IF:2.967)
[T7] Tarcsay, A.; Nyíri, K.; Keserű, G.M., Impact of lipophilic efficiency on compound quality. J Med Chem. 2012, 55, pp. 1252-1260. (IF: 5.248)
Links
References
[1] van de Waterbeemd, H.; Gifford, E., ADMET in silico modelling: towards prediction paradise? Nat Rev Drug Discov. 2003, 2(3), pp. 192-204.
[2] Pan, A.C.; Borhani, D.W.; Dror, R.O.; Shaw, D.E., Molecular determinants of drug-receptor binding kinetics. Drug Discov Today. 2013 Feb 27. pii: S1359-6446(13)00062-7. doi: 10.1016/j.drudis.2013.02.007. [Epub ahead of print]
[3] Hopkins, A.L.; Groom, C.R., The druggable genome. Nat Rev Drug Discov. 2002, 1(9), pp. 727-730.
[4] Antitagets. Roy J Vaz, Thomas Klaubinde (Szerk.), Wiley, 2008, Weinheim.
[5] Lusher, S.J.; McGuire, R.; Azevedo, R.; Boiten, J.W.; van Schaik, R.C.; de Vlieg, J., A molecular informatics view on best practice in multi-parameter compound optimization. Drug Discov Today. 2011, 16, pp. 555-68.
[6] Cheshire, D.R., How well do medicinal chemists learn from experience? 2011, Drug Discov Today. , 16, pp. 817-21.
[7] Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J., Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001, 46 (1-3), 3-26.
[8] Williams, P. A.; Cosme, J.; Ward, A.; et al., Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature 2003, pp. 424, 464-8
[9] Aller, S.G.; Yu, J.; Ward, A. et al., Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science, 2009, 323(5922), pp. 1718–1722.
[10] McGaughey, G.B.; Sheridan, R.P.; Bayly, C.I.; Culberson, J.C.; Kreatsoulas, C.; Lindsley, S.; Maiorov, V.; Truchon, J.F.; Cornell, W.D., Comparison of topological, shape, and docking methods in virtual screening. J Chem Inf Model, 2007, 47, pp. 1504–1519.
[11] Gleeson, M.P., Generation of a set of simple, interpretable ADMET rules of thumb. J Med Chem. 2008, 51 (4), pp. 817-834..
[12] Waring, M.J., Lipophilicity in drug discovery. Expert Opin Drug Discov. 2010, 5 (3), pp. 235-48.
[13] Leeson, P. D.; Springthorpe, B., The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6: 881-890.
[14] Keserű, G. M.; Makara, G. M., The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug Discovery 2009, 8: 203-212.
[15] Schlichting, I.; Berendzen, J.; Chu. K. et al., The catalytic pathway of cytochrome p450cam at atomic resolution. Science 2000, 287, pp. 1615-1622.
[16] Lowe J., F1000Prime Recommendation of [Tarcsay A et al., J Med Chem 2012, 55(3):1252-60]. In F1000Prime, 24 Jan 2012; DOI: 10.3410/f.13484092.14861219. F1000Prime.com/13484092#eval14861219
[17] Kawasaki, Y.; Freire, E., Finding a better path to drug selectivity. Drug Discov Today. 2011, 16, pp. 985-990