|
|
BMe Research Grant |
|

George A. Olah Doctoral School of Chemistry and Chemical Technology
Richter Gedeon Plc.
Supervisor: Dr. György M. Keserű
In silico methodologies aiding fragment-based drug discovery
Introducing the research area
Fighting and preventing diseases are the primary goals of pharmaceutical research. However, the drug discovery process is lengthy and costly and it is burdened by high attrition rates [1]. Fragment-based drug discovery (FBDD) is a recently emerged and quickly spreading technology due to its speed and inherent ability to control the various parameters important in the multi-dimensional optimization process of drug discovery. It seems to be capable of reducing attrition and providing drug candidates even for new biological targets that were previously thought to be intractable [2]. Computer-aided methods can be effectively used in all phases of FBDD aiding medicinal chemistry work and to increase its efficiency [3]. My aim was to evaluate existing and develop new virtual screening methodologies for fragment hit identification and computational methodologies applicable for finding starting points for compound design by fragment linking with a focus on the G protein-coupled receptor (GPCR) protein family, which has a very high relevance to medicinal industry.
Brief introduction of the research site
Gedeon Richter Plc. is a Hungarian headquartered specialty pharmaceutical company. Besides manufacturing generic products, it features one of the largest Central European original research site conducting research of gynecological and central nervous system diseases. Its newest medicine, Cariprazine, which is currently under registration, was effective in phase III clinical trials in schizophrenia and mania indications. My work was carried out in the computational chemistry group in the Lead Discovery Laboratory of the company.
History and context of the research
Fragment-based drug discovery involves the selection, screening and optimization of molecular fragments. Fragments are polar compounds of low molecular weight and low complexity [4] that are able to make optimal interactions with the protein target, thus being better starting points for medicinal chemistry optimization as opposed to lead-like or drug-like molecules identified in high-throughput screening (Fig. 1). A further benefit of fragment-sized molecules is the better representation of chemical space compared to drug-like molecules. However, due to the intrinsically lower affinity of fragments specialized biophysical screening methods with usually lower throughput and a different mindset in their optimization are needed.
Figure 1. A low quality HTS hit (left) and high quality fragment hits (right) [2]
As experimental fragment screening usually faces throughput limitations, efficient methods for virtual fragment screening are also needed in order to enrich biologically active fragments in the screened library. Molecular docking is the usual method of choice for virtual fragment screening that can predict experimental binding modes of fragment ligands [5]. In literature, successful virtual fragment screening campaigns have been demonstrated for soluble proteins and recently against G protein-coupled receptors as well [6,7].
GPCRs are a family of membrane embedded proteins featuring a seven-transmembrane alpha-helical fold and recognizing a wide variety of interacting partners outside the cell from ions to small molecules, lipids and peptide hormones. Recent elucidation of GPCR structures made it possible to apply structure-based computational modeling to this important pharmaceutical target family.
The efficiency of structure-guided fragment optimization is exemplified by Vemurafenib, the first approved drug discovered using FBDD (Fig. 2), which was marketed in 2011 and its idea-to-approval time was only 6 years [8].
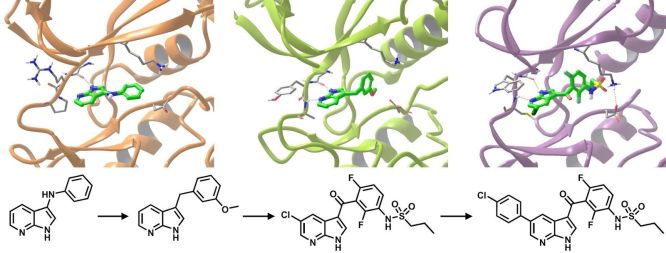
Figure 2. Optimization of Vemurafenib from the azaindole fragment hit by growing [8]
The research goal, open questions
In the first part of the research my aim was to evaluate different protocols for fragment virtual screening on GPCR targets. Since structural information on GPCRs is still restricted to a few targets, the use of experimentally determined structures as well as homology models (modeled structures based on amino acid sequence and tertiary structure similarity) in virtual small molecule and fragment screening was evaluated. The effect of incorporating protein conformational flexibility into both methodologies was also studied and compared to using a single structure for virtual screening. The obtained results were used in prospective fragment screening and the identified fragment hits could be used in a further work as starting points for medicinal chemistry optimization.
In the second part of the research my aim was to study computational methodologies applicable for finding starting points for fragment linking since by linking the two moieties the affinity of the starting fragments might be increased with little synthetic effort [9]. Therefore, besides primary site (or ‘hot spot’) fragment screening I evaluated the performance of a sequential docking protocol for identifying fragments bound in possible secondary sites of proteins. Taking second-site fragment screening forward I have used the protocol to identify fragments for linking inside the binding pocket of the D3 dopamine receptor and also assessed the selectivity of the synthesized compounds against the D2 dopamine receptor on a structural basis.
Methods
One of the most important demands on computational methods used in drug discovery is speed since usually hundreds or hundreds of thousands of compounds need to be evaluated in a prediction model. In exchange for their speed, naturally their precision is bound to be limited. Therefore, a central concept in computer-aided drug design is enrichment, which expresses the ratio between the efficiencies of examining only the compounds predicted to be suitable in some respect and examining the same number of compounds chosen randomly from a library.
Due to their size, proteins and protein-ligand complexes can only be studied using classical mechanical or empirical computational methods. In homology modeling the structure of a protein is modeled based on the known structure of another protein having sufficiently similar amino acid sequence using empirical rules and molecular mechanical conformational searching and optimization. The atomistic description of the flexibility of proteins can be achieved using the similarly force field-based method of molecular dynamics (MD) simulation (Fig. 3). During MD, the time evolution of the system is tracked by calculating the forces acting in the system and solving the classical mechanical equations of motion at a regular time interval.
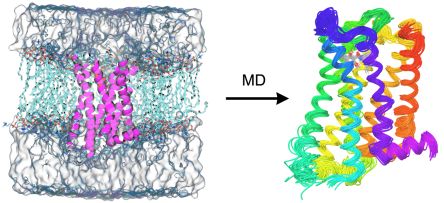
Figure 3. Membrane embedded and solvated structure of the D3 dopamine receptor (left) and the structural ensemble obtained from molecular dynamics simulation (right)
Protein-ligand - in this case fragment - interactions can be modeled using molecular docking, which aims at predicting both the binding mode and the binding free energy. Force field-based conformational searching of the fragments is carried out in the space confined by the protein binding site. Docking can be done in a rigid binding site approximation, a conformational ensemble (obtained e.g. from molecular dynamics simulation) or it is possible to use induced fit methods (IFD) taking into account the conformational flexibility of both the protein and the ligand. The performance of docking-based virtual screening can be characterized by enrichment factors in a retrospective case and the experimentally validated hit rate in a prospective case.
Characterization and comparison of the fragment hits can be done using ligand efficiency metrics such as the generally accepted LE (the contribution of each heavy atom to the binding free energy), LLE (the separation between binding affinity and logP, the water-octanol partition coefficient) and LELP (the ratio of logP and LE). The latter so called lipophilic efficiency indices were shown to be predictive of pharmacokinetic and safety parameters of the compounds [10].
In my work I used the Schrödinger software suite for modeling. The Prime module was used for homology modeling of GPCRs with yet undetermined structures and the Glide quick docking module was used for docking of the fragment ligands. The associated Python API scripting environment was used for the automation of sequential docking. The NAMD software was used in a collaboration for molecular dynamics simulations and ChemAxon cheminformatics tools were used for calculating molecular parameters and handling compound libraries.
Results
Primary binding site virtual screening
Homology models of the H4 histamine and 5HT6 serotonin receptors were constructed and also X-ray structures of the D3 dopamine and CXCR4 chemokine receptors were used as starting structures in molecular dynamics simulations. Docking-based retrospective enrichment studies were carried out on the starting structures and on the obtained conformational ensembles and were characterized by enrichment factors (EF) and by the area under the receiver operating characteristic curves (AUC). As can be seen from Fig. 4, among the starting structures only the H4 homology model was able to separate active and decoy molecules and the best single structures from MD were always superior to the initial models, regardless of the target and the evaluation method (EF or AUC) [T1].
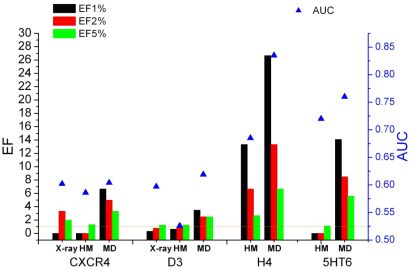
Figure 4. Enrichment factors (colored bars) and AUC values (blue triangles) for the CXCR4, D3, H4, and 5HT6 structures obtained with X-ray, homology model (HM), and the best molecular dynamics frame (MD)
Next, prospective virtual fragment screening of the Gedeon Richter fragment library was performed against the available structural models of the dopamine D3 receptor and the histamine H4 receptor. Approximately 50-50 fragments were selected for biological testing both from single structure and ensemble docking. With the different protocols validated hit rates of 16-32% were achieved and novel fragment hits with favorable LE (greater than 0.3) and LELP (lower than 10) values were identified (Fig. 5). Both X-ray structure and homology model were capable of providing useful hits in virtual screening. The superiority of the ensemble docking approach was not witnessed but it gave different hit sets and thus the two methods were complementary [T2].
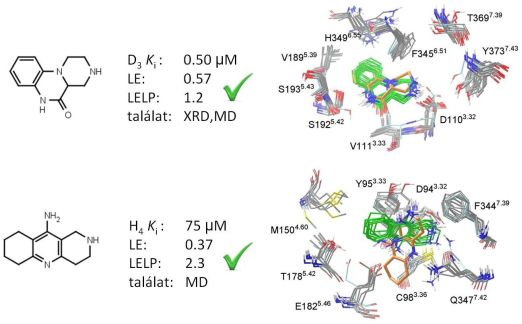
Figure 5. Novel fragment hits from prospective fragment screening, their binding affinities, ligand efficiency indices and predicted binding modes from single structure docking (orange) and docking to the structural ensembles (green)
Secondary binding site virtual screening
In the next part of the work, performance of a sequential docking protocol was evaluated for modeling cooperatively bound ligands in the binding sites of 129 ternary and higher order protein-small molecule complexes and then 32 protein-fragment complexes. From the small molecule data set it was seen that docking of two ligands is successful in 55% of the cases while docking of more ligands generally failed. When considering only a subset with drug-like ligands bound in closed binding sites, the success rate increased to 68% with the best investigated protocol. For the five structures of the HSP90 protein crystallized with fragment ligands all docking steps were successful, which motivated the evaluation of the protocol for the new fragment data set [T3].
As can be seen from Fig. 6, docking was successful for all ligands: a pose was always found with atomic RMSD < 2.0 Å to the experimental binding conformation. Furthermore, in 77% of the cases a pose with atomic RMSD < 1.0 Å was found. High docking accuracy is especially important for virtual linker design in fragment optimization. Similarly good results were obtained for cross-docking, i.e. when fragment pairs were docked to different X-ray structures of the same protein [T4].
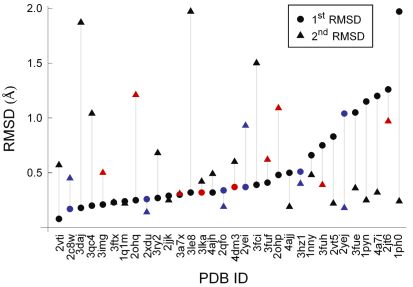
Figure 6. Multiple fragment docking results obtained by sequential docking. Plot markers are colored red for scoring errors and blue for complexes where structural waters were included
Finally, the sequential docking methodology was applied for fragment docking and linking to the D3 crystal structure and a D2 homology model since selective D3 antagonists or partial agonists are investigated in the treatment of schizophrenia, depression and bipolar mania. Docking a focused library of basic fragments, an aryl-piperazine fitting to the primary binding site was identified. Among the fragments docked to the secondary binding site a cyclohexylaminosulfonamide fragment produced a robust binding mode featuring an H-bond with Tyr1.39 that indicated selectivity between the two receptors (Fig. 7). Three linked molecules were synthesized and indeed, when this fragment was linked to the primary site aryl-piperazine, the resulting compound showed high affinity and 55-fold selectivity towards the D3 receptor [T5].
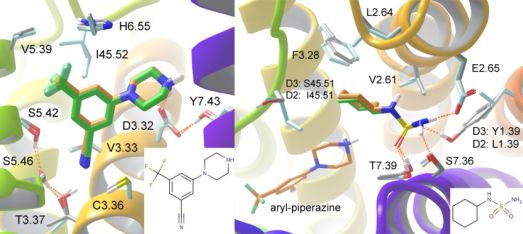
Figure 7. Binding modes of the top primary (left) and secondary site fragments (right). D3 and D2 binding sites are overlaid in grey and light blue carbons respectively and docked poses of the ligands in orange and green carbons, respectively
Expected impact and further research
The publication about GPCR enrichment factors [T1] achieved 3rd place among the most read JCIM articles, and the article describing prospective GPCR fragment screening [T2] was downloaded almost 500 times in one month after publication, showing the interest towards these papers. Multiple fragment docking was investigated by only a few groups [11] but the fact that the paper on D3 fragment linking [T5] was accepted in ACS Med Chem Letters also showed the interest towards the applications.
The demonstrated results can be used in the design of fragment screening campaigns against further GPCRs. The identified fragment hits of D3 and H4 serve as suitable starting points for medicinal chemistry optimization. Sequential fragment docking can also be used against further target proteins and it was proposed to aid X-ray structure elucidation when fragments are screened in cocktails [12].
Publications, references, links
Publications
T1. Tarcsay, Á., Paragi, G., Vass, M., Jójárt, B., Bogár, F., Keserű, G. M.: The impact of molecular dynamics sampling on the performance of virtual screening against GPCRs. J. Chem. Inf. Model., 2013, 53, pp. 2990-2999 (IF: 4.304)
T2. Vass, M., Schmidt, É., Horti, F., Keserű, G. M.: Virtual fragment screening on GPCRs: a case study on dopamine D3 and histamine H4 receptors, Eur. J. Med. Chem., 2014, 77, pp. 38-46 (IF: 3.499)
T3. Vass, M., Tarcsay, Á., Keserű, G. M.: Multiple ligand docking by Glide: implications for virtual second-site screening, J. Comput. Aided Mol. Des. 2012, 26, pp. 821-834 (IF: 3.172)
T4. Vass, M., Keserű, G. M.: Fragments to link. A multiple docking strategy for second site binders, MedChemComm 2013, 4, pp. 510-514 (IF: 2.722)
T5. Vass, M., Ágai-Csongor, É., Horti, F., Keserű, G. M.: Multiple fragment docking and linking in primary and secondary pockets of dopamine receptors, ACS Med. Chem. Lett. Accepted (IF: 3.311)
References
1. Leeson, P.D., St-Gallay, S. A.: The influence of the 'organizational factor' on compound quality in drug discovery, Nat. Rev. Drug Discov. 2011, 10, 749
2. Rees, D.C., Congreve, M., Murray, C. W., Carr, R.: Fragment-based lead discovery, Nat. Rev. Drug Discov. 2004, 3, 660
3. Sheng, C., Zhang, W.: Fragment informatics and computational fragment-based drug design: an overview and update, Med. Res. Rev. 2013, 33, 554
4. Congreve, M., Carr, R., Murray, C., Jhoti, H. A 'rule of three' for fragment-based lead discovery? Drug Discov. Today 2003, 8, 876.
5. Sándor, M., Kiss, R., Keserű, G. M.: Virtual fragment docking by Glide: a validation study on 190 protein-fragment complexes, J. Chem. Inf. Model. 2010, 50, 1165
6. de Graaf, C., Kooistra, A.J. et al.: Crystal structure-based virtual screening for fragment-like ligands of the human histamine H1 receptor, J. Med. Chem. 2011, 54, 8195
7. Chen, D., Ranganathan, A. et al.: Complementarity between in silico and biophysical screening approaches in fragment-based lead discovery against the A2A adenosine receptor, J. Chem. Inf. Model, 2013, 53, 2701
8. Tsai, J., Lee, J.T. et al.: Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity, Proc. Natl. Acad. Sci. USA 2008, 105, 3041
9. Ichihara, O., Barker, J., Law, R. J., Whittaker, M.: Compound design by fragment-linking, Mol. Inf., 2011, 30, 298
10. Hopkins, A.L., Keserű, G.M. et al. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov., 2014, 13, 105.
11. Hoffer, L., Horvath, D.: S4MPLE - sampler for multiple protein-ligand entities: simultaneous docking of several entities, J. Chem. Inf. Model, 2013, 53, 88
12. Nair, P.C., Malde, A. K., Drinkwater, N., Mark, A. E.: Missing fragments: detecting cooperative binding in fragment-based drug design, ACS Med. Chem. Lett., 2012, 3, 322
Links
Fragment based drug discovery blog