|
|
BMe Research Grant |
|

Kollár Levente
BMe Research Grant - 2022
![]()
George A. Olah Doctoral School of Chemistry and Chemical Technology
BME Faculty of Chemical Technology and Biotechnology, Department of Organic Chemistry and Technology Research Centre for Natural Sciences, Medicinal Chemistry Research Group
Supervisor: Dr. Keserű György Miklós
Development of covalent inhibitors against various drug targets
Introducing the research area
Some active ingredients that act as covalent inhibitors have been part of our drug supply for many decades, yet research on molecules that follow this mechanism of action has only begun to gain ground around the turn of the millennium. This is because their use may even carry risks, but over the past decade, researchers have begun to recognize and appreciate the invaluable therapeutic potential inherent in them. With no claim of being exhaustive, the advantages of covalent inhibitors include their high potency, long residence time, good biochemical utilization, and their ability to target proteins that cannot be targeted by purely non-covalent interactions. In our work, we aimed to identify and develop covalent inhibitors against several therapeutically important proteins.
Brief introduction of the research place
The present research takes place in the Research Centre for Natural Sciences (TTK), part of the Eötvös Loránd Research Network (ELKH), in the Medicinal Chemistry Research Group headed by Dr. György Miklós Keserű, corresponding member of the Hungarian Academy of Sciences and a full professor. The group has an extensive network of national and international contacts, and we are interested in several drug research programs within the framework of the established collaborations. We have been in the research of covalent inhibitors since 2015.
History and context of the research
Covalent inhibitors are compounds that exert their effects by forming a covalent bond with specific nucleophilic amino acid residues of the target proteins. Looking at the history of drug discovery, we can state that these compounds have been kept at a distance for a long time. This is understandable in some respects, as the use of high-reactivity covalent inhibitors is associated with an increased risk of immunogenicity and toxicity compared to drugs with non-covalent mechanisms of action [1,2].
However, the situation is nuanced by the fact that, for example, aspirin, one of our oldest safe drugs, acts as a covalent inhibitor, and β-lactam antibacterial agents, the first representative of which is penicillin, also follow this mechanism of action (Figure 1). The paradigm shift can be traced back to the early 2000s when drug researchers increasingly recognized and began to address the potential of covalent inhibitors in the right place. The process of covalent inhibition follows a two-step mechanism: the first step is molecular recognition, in which the ligand is stabilized by forming non-covalent interactions at the binding site, followed by the formation of a covalent bond with the particular amino acid residue. This results in a great advantage, the outstanding efficacy: complete inhibition can be achieved, and the therapeutic effect can typically be induced by using a lower dose. As a result of non-equilibrium binding, their use is less sensitive to pharmacokinetic characteristics. It should also be emphasized that the covalent approach is also a weapon for target proteins where the binding site is on the surface or highly flexible and cannot be targeted by the formation of non-covalent interactions alone [3].
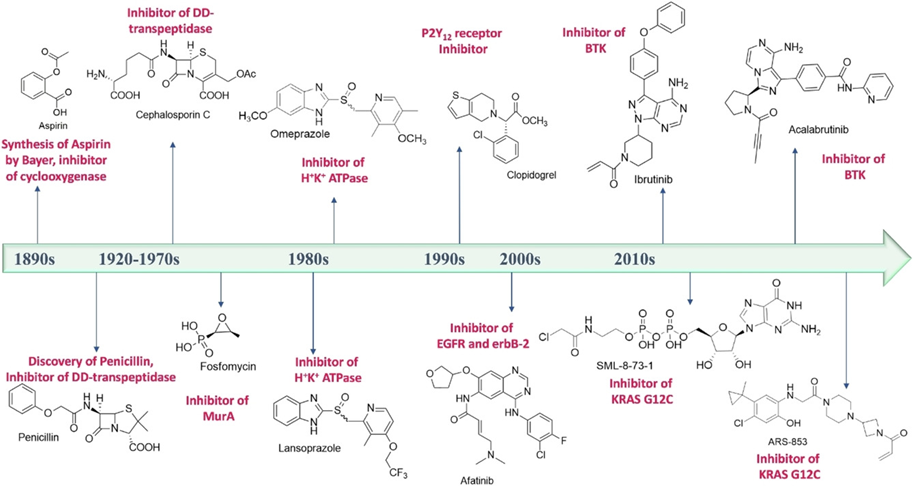
Figure 1: The most important covalent inhibitors currently in use, the order of their discovery can be traced on the timeline [4].
The research goals, open questions
In our work, we aimed to develop covalent inhibitors for different protein targets, thus demonstrating the versatility of the approach.
We chose the immunoproteasome (iPS) as our main target, a multicatalytic protein complex, which is responsible for the breakdown of damaged and poorly folded proteins, and its dysfunction has been linked to several oncological, autoimmune, and neurodegenerative diseases. Different immunoproteasome subunits have different catalytic activities, and their selective inhibition is desirable to reduce potential side effects. Due to the structural similarity of the subunits, the identification of selective inhibitors is challenging, however, the β5i subunit allows covalent targeting due to the specific cysteine amino acid present at its binding site. In our work, we aimed to identify and develop β5i-selective immunoproteasome inhibitors [5,6].
Penicillin-binding proteins (PBPs) are enzymes that are primarily involved in the peptidoglycan biosynthesis of bacteria, thereby playing a key role in the process of building the bacterial cell wall. Inhibition of penicillin-binding proteins is typically accomplished with β-lactam-type compounds (e.g. penicillin), however, the development of bacterial resistance is an increasing problem to be addressed, and research and development of additional antibacterial agents is desirable. We aimed to identify new antibacterial agents in which the reactive group responsible for the formation of a covalent bond is not the conventional β-lactam but another serine-reactive warhead [7,8].
The COVID-19 epidemic has fundamentally changed the world. Overcoming the pandemic has become a common interest and task of all mankind. It was soon established that the main protease of coronavirus (3CLPro) plays a key role in viral replication, and its inhibition proved to be a beneficial strategy, making it one of the number one drug targets. The mentioned protein is a cysteine protease and is therefore suitable for targeting with covalent inhibitors [9,10].
Methods
We carry out the mentioned drug research projects together with our national and international partners. Our group is responsible for supporting the work with a computational chemical background, as well as for the synthesis of molecules obtained as a result of computer-aided drug design, and for the analytical and biophysical characterization of these and their protein complexes. For all three target proteins, we are working with researchers from the University of Ljubljana to investigate the biochemical activity of the compounds. In the research of covalent inhibitors, it is essential to prove the binding of the compounds to the protein, in which mass spectrometric measurements play a key role, with the help of the TTK MS Metabolomics Research Laboratory. If the molecule is shown to be able to label the protein, digestion of the resulting conjugate with appropriate enzymes can provide information on exactly which amino acid residue the small molecule has bonded to.
An additional difficulty in research on immunoproteasome inhibitors, in addition to the selectivity challenges already mentioned, is that a large proportion of inhibitors known in the literature have a peptide or peptidomimetic structure, which can often result in poor metabolic stability or low oral bioavailability. To overcome this, fragment screening was performed to measure the activity of hundreds of fragment-sized molecules (small, simple, polar molecules) at a suitable concentration against the β5i subunit of the immunoproteasome, and to determine the IC50 for the most active compounds.
Compound libraries were constructed based on the structures that proved to be active, and these were investigated in the context of structure-activity relationships to give three analogous families: benzoxazole-2-thione, benzimidazole-2-thione, and benzothiazole-2-thione (together: benzXazoles), which were further optimized. A halogen scan was performed to examine the preferred directions for the chemical modification of the fragments, which led to the purchase and synthesis of dozens of compounds. The proper purity of the purchased compounds was checked by HPLC and 1H NMR and further purified if necessary. Almost all the synthesized compounds were unknown in the literature, so complete characterization (HPLC, 1H NMR, 13C NMR, HRMS, melting point) was performed in each case. Biophysical methods and biochemical tests were used to investigate the mechanism of inhibition.
In the research of new antibacterial agents (PBP1b inhibitors), a library containing several serine-binding structural elements was subjected to a biochemical assay: carbamates, boronic acids, boronic esters, and various N-methylated heterocycles were tested. It was an obstacle that interference occurred with the components of the biological test in relation to the cysteine binding of the compounds, so a new, appropriate protocol was developed. Calculations were performed to elucidate the binding mechanism of covalent inhibitors and to identify the structural elements and properties that determine affinity. Several boronic acids were synthesized, which could fill multiple binding pockets simultaneously, thereby creating strong inhibition, according to modeling.
Due to the great international interest, a large amount of data (e.g. X-ray crystallographic structure) on the interaction of 3CLPro with small molecules has been generated in a short time that can be used for the rational design of new 3CLPro inhibitors. In our work, we try to design and synthesize effective new 3CLPro inhibitors using new methodological solutions (e.g. fragment coupling based on experimental binding methods). In addition, by screening our compounds previously tested against other protein targets (e.g. benz-X-azole library), we were able to identify new 3CLPro inhibitors whose mechanism of action we would like to interpret by high-level molecular modeling methods.
Results
As a result of fragment screening (Figure 2), we identified three families of related compounds that were found to be inhibitors of the β5i subunit of the immunoproteasome with high ligand efficiency.
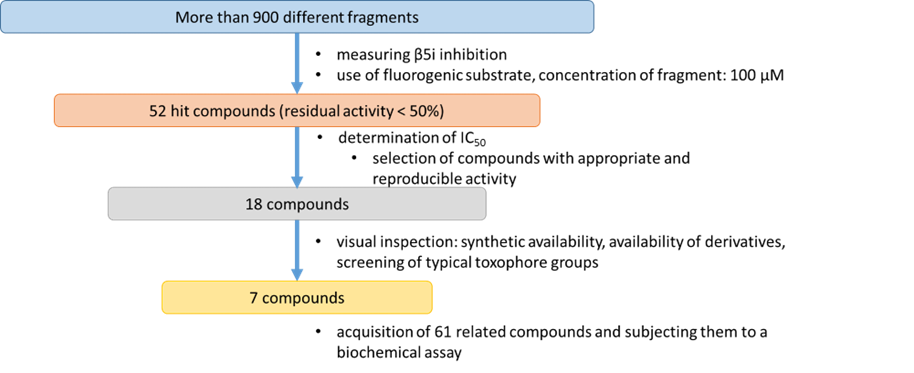
Figure 2: Process of fragment screening
Following the fragment-based drug discovery strategy, these compounds with benz-X-azole core were first investigated as chloro-monosubstituted derivatives of the backbones (chloro-scan). Given that the most potent inhibition was shown by the 6- and 7-substituted analogs, such derivatives were further prepared and subjected to biochemical testing (Figure 3). The activity of benzimidazole-2-thione compounds and their N-methylated analogs was also compared. The latter were typically much more potent, so a number of N-methylbenzimidazole-2-thione derivatives were synthesized and tested. Another series has been prepared in which the 6-chlorobenzimidazole-2-thione backbone carries typically small, polar substituents on the nitrogen atom. A total of 68 derivatives were synthesized [S1].
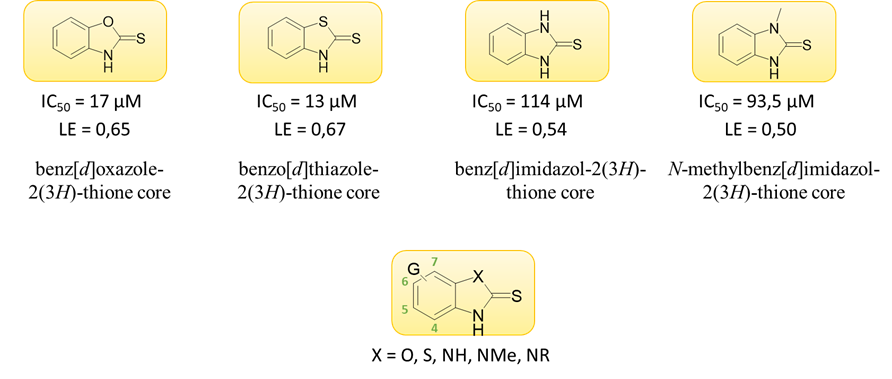
Figure 3: Structure of the identified scaffolds in the company of biological activity and ligand efficiency (top); the general formula of the synthesized derivatives, the numbering of the backbone is indicated (bottom)
Optimization of the warhead was also performed by replacing the thione group uniformly present in the identified compounds with other cysteine-reactive moieties. As a result, we identified carbonitrile derivatives that were more advantageous than thiones in terms of overall activity, selectivity, and reactivity. A 27-membered systematically substituted library of benzoxazole-2-carbonitrile compounds was constructed (Figure 4), and the stability of the compounds and their reactivity against model compounds (glutathione, N-acetyl cysteine) was tested. Their biological activity against the β5i subunit of the immunoproteasome was determined, and selectivity was also examined for the most potent compounds. Group efficiency analysis was performed to examine the most preferred positions for substitution in the backbone.
In our studies, we searched for a correlation between the reactivity of the compounds and their biochemical activity, but to some surprise, we did not find a trend between them. This fact also highlights that non-covalent recognition is also a key issue for covalent inhibitors of fragment size [S2].
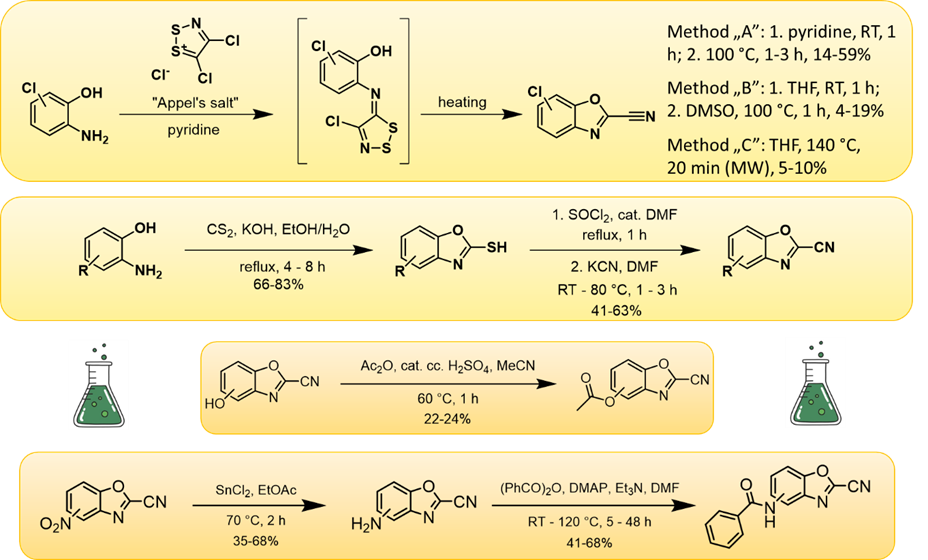
Figure 4: Reactions commonly used to construct the benzoxazole-2-carbonitrile compound library
The most promising compounds had IC50 values (β5i) between 2-4 μM. We have also dealt with the synthesis of molecules that contain the (R) -boroleucine moiety found in Bortezomib and the benzoxazole-2-carbonitrile or benzimidazole-2-carbonitrile backbone connected with a linker: as a result, possibly they can act as “bidentate” (able to form two covalent bonds) compounds (Figure 5). Twelve derivatives were synthesized (Figure 6), and a submicromolar IC50 (β5i subunit) was reached for the most potent molecule [S2].
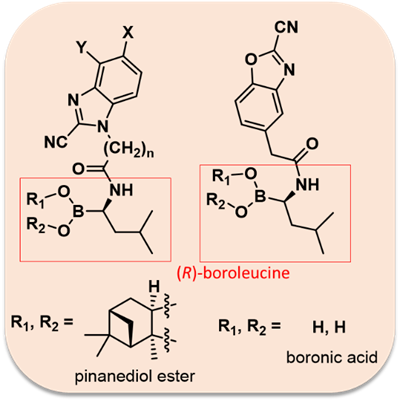
Figure 5: General structure of "bidentate" compounds
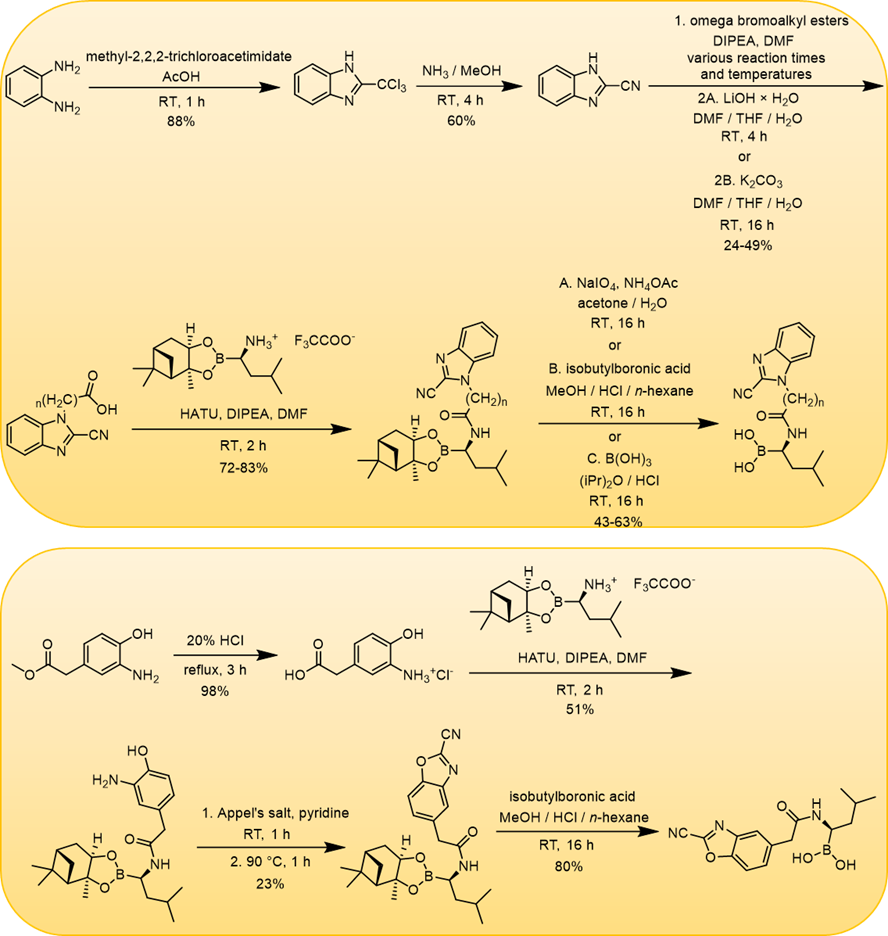
Figure 6: Synthesis of “bidentate” compounds
Attempts have been made to identify inhibitors that bind to the β5i subunit of the immunoproteasome from other approaches. 404 commercially available 5- and 6-membered cyclic boronic acids were subjected to virtual screening. Twelve of the best-performing compounds were tested in a biochemical assay and had measurable activity, being the first cyclic boronic acid immunoproteasome inhibitor to be described in the literature [S3].
In the framework of the research of PBP1b inhibitors, our partners have developed a new biochemical assay, which can also be used to study cysteine-reactive compounds. In addition, two boronic acids with significant activity (IC50 values: 77 µM and 50 µM) were successfully synthesized and identified. These compounds can serve as a starting point for further optimization experiments.
The previously prepared benz-X-azole library was tested against the main protease of coronavirus (3CLPro) and several inhibitors were identified. Based on the obtained results, additional derivatives were synthesized, and the mechanism of action of inhibitors was further studied by computational chemical methods.
Expected impact and further research
Further optimization of benzoxazole-2-carbonitriles, which have been extensively studied in the research of immunoproteasome inhibitors, is desirable in light of the results obtained so far. The planned fragment growth campaign is based on the group impact analysis performed. Although the most active members of the bidentate compounds are not considered to be selective (pan-proteasome inhibitors), their development as cancer therapeutic agents may be rational.
The results of the research on PBP1b inhibitors so far are planned to be published soon, and an even wider screening of possible warheads will be carried out. Another option lies in the optimization of boronic acids.
A paper investigating the mechanism of action of benz-X-azole-based inhibitors identified for the 3CLPro protein is under preparation and will be ready for submission to the publisher soon. Our research is currently underway to design and synthesize new 3CLPro inhibitors by linking fragments, which we plan to continue.
Publications, references, links
List of corresponding own publications
[S1] Kollár, L., Gobec, M., Szilágyi, B., Proj, M., Knez, D., Ábrányi-Balogh, P., Petri, L., Imre, T., Bajusz, D., Ferenczy, G. G., Gobec, S., Keserű, G. M., Sosič, I. „Discovery of selective fragment-sized immunoproteasome inhibitors”, Eur. J. Med. Chem., 2021, 219, 113455. (IF: 6.514)
[S2] Kollár, L.; Gobec, M.; Proj, M.; Smrdel, L.; Knez, D.; Imre, T.; Gömöry, Á.; Petri, L., Ábrányi-Balogh, P.; Csányi, D.; Ferenczy, G. G.; Gobec, S.; Sosič, I.; Keserű, G. M. “Fragment-Sized and Bidentate (Immuno)Proteasome Inhibitors Derived from Cysteine and Threonine Targeting Warheads”, Cells, 2021, 10, 3431. (IF: 6.600)
[S3] Kollár, L., Ferenczy, G. G., Proj, M., Gobec, M., Gobec, S., Sosič, I., Keserű, G. M. “Virtual Screening and Biochemical Testing of Borocycles as Immunoproteasome Inhibitors”, Per. Pol. Chem. Eng., 2021, 65(3), 292–298. (IF: 1.257)
Table of links
Medicinal Chemistry Research Group
main protease of coronavirus (3CLPro)
List of references
[1] Singh, J., Petter, R. C., Baillie, T. A., Whitty, A. Nat. Rev. Drug Discov. 2011, 10, 307–317.
[2] Bauer, R. A., Drug Discov. Today 2015, 20, 1061–1073.
[3] Johnson, D.S., Weerapana, E., Cravatt, B.F. Future Med. Chem. 2010, 2, 949–964.
[4] Ghosh, A. K., Samanta, I., Mondal, A., Liu, R. ChemMedChem, 2019, 14, 889–906.
[5] Murata, S., Yashiroda, H., Tanaka, K. Nat. Rev. Mol. Cell Biol., 2009, 10, 104–115.
[6] McCarthy, M. K., Weinberg, J. B. Front. Microbiol., 2015, 6, 21.
[7] Inglis, S. R., Strieker, M., Rydzik, A. M., Dessen, A., Schofield, C. J., Anal. Biochem. 2012, 420, 41–47.
[8] Zervosen, A., Sauvage, E., Frère, J. M., Charlier, P. Luxen, A. Molecules 2012, 17, 12478–12505.
[9] Hoffman, R. L., Kania, R. S., Brothers, M. A., Davies J. F., Ferre, R. A., Gajiwala, K. S., He, M., Hogan, R. J., Kozminski, K., Li, L. Y., Lockner, J. W., Lou, J., Marra, M. T., Mitchell, L. J., Murray, B. W., Nieman, J. A., Noell, S., Planken, S. P., Rowe, T., Ryan, K., Smith, G. J., Solowiej, J. E., Steppan, C. M. Taggart B. J. Med. Chem. 2020, 63(21), 12725–12747.
[10] Mody, V., Ho, J., Wills, Mawri, A., Lawson, L., Ebert, M. C. C. J. C., Fortin, G. M., Rayalam S. Commun. Biol. 2021, 4, 93.