|
|
BMe Research Grant |
|

George A. Olah Doctoral School of Chemistry and Chemical Technology
Department of Inorganic and Analytical Chemistry
Supervisor: Dr. Viola Horváth
Selective protein-recognition with molecularly imprinted polymers
Introducing the research area
Molecular imprinting creates polymers capable of selectively recognizing and binding a target compound of choice. The success of imprinting small target molecules is demonstrated by the fact that molecularly imprinted polymer (MIP) sorbents are readily available on the market. The imprinting of larger molecules (e.g. proteins), however, is still in its infancy. I develop novel methods to overcome protein imprinting related difficulties with the aim of applying the MIPs as a protein-sensor.
Brief introduction of the research place
The Research Group of Pharmacokinetics of the Departments of Inorganic and Analytical Chemistry has been researching molecular imprinting since the turn of the millennium. Along with the development of selective sorbents for small molecules [1-4], the group also focuses on theoretical description of the analytical performance of molecularly imprinted polymers (MIPs) [5-9]. Moreover, by developing novel polymerisation techniques the group is in the forefront of the fast optimisation of MIPs [10-11], the preparation of spherical polymer particles for chromatographic applications [B1, 12], as well as the imprinting of large templates (e.g. proteins) [B2, 13-15].
History and context of the research
Medical practice often requires the measurement of the concentration of certain biomacromolecules to establish a diagnosis or to manage a known condition. Many of these measurements are performed using immunoassays, in which a specific antibody recognizes the compound of interest (antigen) and binds it from the complex sample (e.g. blood). The interaction between antibody and antigen is very strong and selective, but antibodies are easily degraded. More robust “plastic antibodies” can be synthesized using the method of molecular imprinting.
These molecularly imprinted polymers (MIPs) are able to bind a specific target compound with affinity and selectivity comparable to those of the natural antibody-antigen interaction. MIPs are synthesized in the presence of the target molecule which acts as a template: suitably chosen monomers are mixed with the template molecules and allowed to preassemble through covalent or secondary bonds; this arrangement is then secured by the formation of a polymer network. The obtained rigid, highly cross-linked polymer monolith is ground in a mortar and sieved to fine particles. Upon extraction, the template molecules leave behind cavities in the polymer, the shape and arrangement of functional groups of which are complementary to the template (Figure 1). When using the polymer, the target molecules can bind into these cavities, by re-formation of the bonds that were cleaved during template extraction. In order to evaluate the imprinting effect, a reference polymer, named non-imprinted polymer (NIP) is always prepared along with the MIP, in an identical manner except for leaving out the template from the polymerisation mixture.
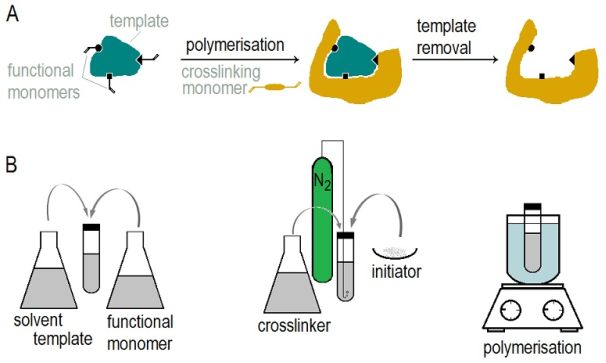
Figure 1: Scheme of molecular imprinting (a)
and the steps of traditional MIP synthesis (b)
Successful imprinting of small molecules has a wide literature in various fields of application (catalysis, sensors, separation, drug delivery), however, the imprinting of proteins is still in its infancy. The reason for this lies in the difficulties arising from proteins’ large size and structural complexity. Traditional bulk polymerisation method is not suitable for imprinting biomacromolecules, since their movement is hindered in the highly cross-linked polymer network, impeding them from accessing the binding sites inside the polymer.
The research goal, open questions
Based on the traditional, highly cross-linked bulk polymerisation method, two strategies have been evolved to imprint proteins, differing in the way of facilitating protein movement in the polymer. One approach is to increase the diffusion constant by significantly reducing the amount of cross-links in the polymer, that is, by preparing polymer gels.
Another approach is to reduce the diffusion length: here, the highly cross-linked structure that better preserves the imprints can be retained, but the imprints are confined to the surface of the polymer to facilitate the free material transport between the sample solution and the polymer phase.
It is noteworthy that surface imprints are also formed during bulk polymerisation but only randomly and to a negligible extent compared to the polymer volume. In order to achieve a MIP with high binding capacity, the imprint formation has to be specifically concentrated to the polymer surface and in addition, a polymer format with high surface area to volume ratio has to be formed. This is best achieved by the preparation of nanostructures.
I have developed a method based on nanosphere lithography to prepare thin polymer films bearing protein imprints on their surface. Avidin, a protein found in egg-white was chosen as a model template. I tethered the protein to sub-micron sized particles and created a compact layer of these beads on a substrate (Figure 2). I filled the voids between the beads with a polymer, in which the imprints of the beads as well as of the attached protein molecules were formed during polymerisation. Finally, by extracting the template (protein and beads) I obtained the imprinted polymer surface.
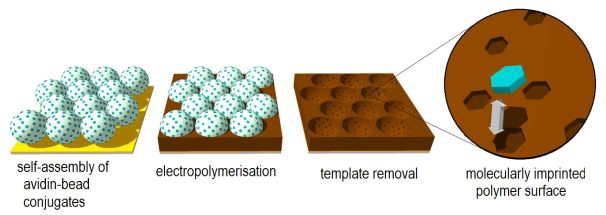
Figure 2: The proposed steps of protein-imprinting based on nanosphere-lithography
Methods
The target protein was attached to Ø750 nm polystyrene latex beads through a cleavable cross-linker (Figure 3). This was necessary because we planned to dissolve the beads in toluene as a means of template removal but the protein precipitated in the organic solvent, preventing complete dissolution of the beads. This problem was overcome by first cleaving the cross-linker (by reduction of its disulphide bond), thereby releasing the protein from the beads’ surface. The bare latex beads could then be instantly dissolved in toluene. Attachment of the protein to the beads was verified by the change in their surface zeta potential.
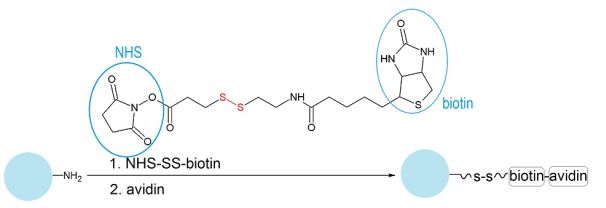
Figure 3: Attachment of avidin to amine-functionalized polystyrene (PS) beads using the avidin-biotin interaction
The beads were assembled into a compact layer on a gold-plated substrate by drop-casting, a common nanosphere-lithographic technique (basically by placing a droplet of the bead suspension on the substrate and letting the solvent slowly evaporate which induces the self-assembly of the nanobeads into a closely packed array). The bead layers were investigated by scanning electron microscopy. The monomer of choice (3,4-ethylenedioxythiophene, EDOT, in poly(sodium 4-styrenesulfonate) electrolyte) was electrochemically oxidised at a constant potential to deposit a polymer layer in the voids of the beads. The morphology of the polymer films after template removal were investigated by atomic force microscopy. The thickness of the polymer was controlled by the amount of electrical charge passed during polymerisation, and was optimised to obtain maximum imprinted to non-imprinted surface ratio. This optimal film thickness, based on geometrical calculations, corresponds to approximately half-height of the beads.
Removal of the template beads revealed areas of bare gold (where the beads touched the substrate). These areas were covered with a self-assembled monolayer of a hydrophilic thiol compound to prevent non-specific adsorption of proteins which would occur on bare gold but not on this layer.
The binding of the target and other proteins to the polymer films was investigated by a quartz crystal microbalance. The polymer-coated quartz crystal was mounted into a flow cell of 120 μl and a buffer solution was passed through the cell at a flow rate of 60 μl/min. 250 μl protein solutions of different concentrations were injected into the cell while monitoring the crystal’s resonant frequency. The amount of bound protein was calculated from the resulting frequency shift using the Sauerbrey-equation.
Results
Protein-modification and assembly of the beads
Since one avidin bears four biotin-binding sites, the step of protein attachment to the biotinylated beads had to be optimised in terms of avidin excess and bead concentration on order to suppress cross-linking of the beads by the protein. Avidin has a high isoelectric point (pI=10.5), meaning that it is positively charged at the applied pH of 7.4. Therefore, avidin’s attachment to the beads shifted their surface zeta-potential to a more positive value (Figure 4).
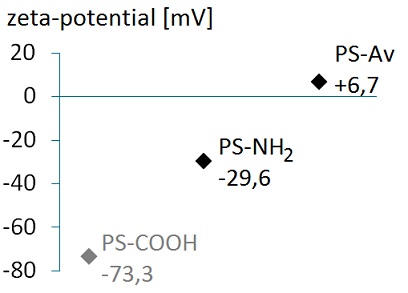
Figure 4: Successful attachment of avidin to the amine-functionalised beads is demonstrated by the positive shift in their surface zeta-potential.
(Please note that the surface pKa of aliphatic amine groups can be as low as 4-4.5 compared to 9-10 in bulk solution phase [16] - this explains the negative value of surface zeta-potential of amine-functionalised beads. For comparison, the surface zeta-potential of carboxylated beads is also shown.)
Amine-functionalised beads (used for the preparation of the non-imprinted polymer) self-assembled into a hexagonally ordered layer upon drop-casting (Figure 5a). In contrast, such compact layer could not be obtained from the protein-modified beads: depositing the amount of beads required for a close-packed monolayer resulted in the formation of loosely packed multilayers on the substrate (Figure 5b). Although compact ordering of the protein-modified beads would have the benefit of increased imprint formation, perfect ordering is not crucial for our application (unlike other applications of nanosphere lithography, such as optical device fabrication).

Figure 5: Scanning electron micrographs of drop-casted layers of (a) reference and (b) avidin-modified beads. Scale bars are 10 µm.
Electrochemical polymerisation, template removal
Atomic force microscopy of the polymer films deposited between the beads using different amounts of charge (Figure 6) clearly demonstrates that the thicker the film, the larger portion of the beads is embedded in the polymer: the depths of the beads’ imprints increase, and their upper diameter increases until it reaches the bead diameter (A-C). Then, as the polymer film outgrows the bead’s “equator”, the upper diameter of the imprints starts to decrease (D). The charge density corresponding to the optimal film thickness (i.e. half-height of the beads) was determined to be 17 mC/cm2.
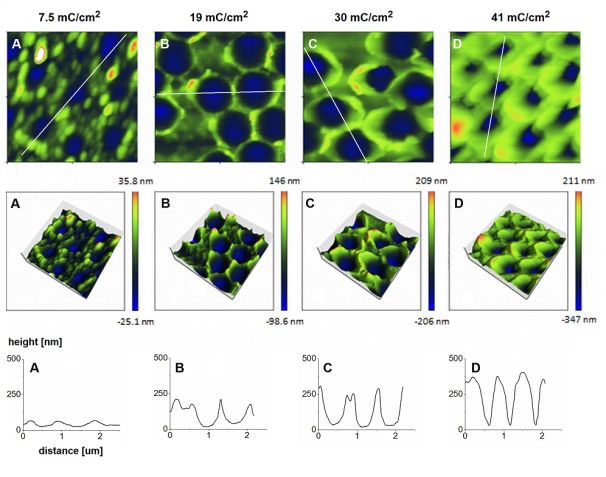
Figure 6: Morphology after template removal of polymer films grown using different amounts of charge - atomic force microscopy images (upper rows) and depth profiles along the lines specified in the first row
Templates were extracted in two consecutive steps: first the disulphide bond of the cross-linker was reduced to cleave the protein off the beads. Then the polymer film was soaked in toluene which easily dissolved the beads (the PEDOT/PSS material was not impaired by toluene). Using non-imprinted polymers, it is demonstrated in Figure 7 that blocking the gold areas (revealed upon template removal) with a hydrophilic monolayer effectively reduces non-specific adsorption of proteins to the surface.
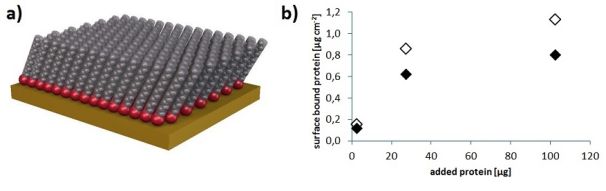
Figure 7: (a) Self-assembled monolayer – the blocking agent (HS-TEG) binds to the gold by its thiol head group (red), while the hydrophilic tail (grey, in our case: a tetra(ethyleneglycol) chain following an alkyl chain of 11 carbons) resist protein-adsorption [17].
(b) Almost 30% less protein was adsorbed on a NIP film treated with HS-TEG (filled diamonds) than on a NIP not blocked after bead removal (empty diamonds)
Protein binding on the polymer films
The effect of imprinting with avidin is demonstrated in Figure 8: while the PEDOT/PSS material itself (non-imprinted polymer) can only bind 0.21 μg/cm2 of avidin, its binding capacity increased 6.5-fold due to the formation of specific imprints. This so-called imprinting factor is among the highest in the literature of protein-imprinted polymers.
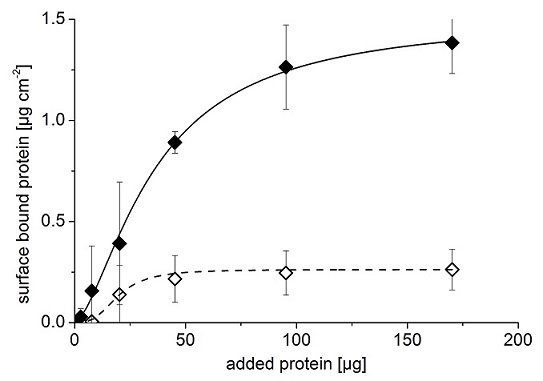
Figure 8: Binding of avidin to imprinted (filled diamonds) and non-imprinted (empty diamonds) polymers
Selectivity of the binding sites was investigated by measuring the adsorption of different proteins on the polymer films. Figure 9 shows that deglycosylated forms of avidin (Neutravidin - NA, Extravidin - EA) as well as another biotin-binding protein whose structure is almost identical to that of avidin (Streptavidin - SA), are only bound by the polymers to a negligible extent. Bovine serum albumin (BSA), a protein similar in size to avidin, was also poorly retained by the films.
However, significant adsorption was observed on both NIP and MIP films in the case of lysozyme (Lys), a small protein of similar isoelectric point to that of avidin. This demonstrates that electrostatic interactions play an important role in the recognition process.
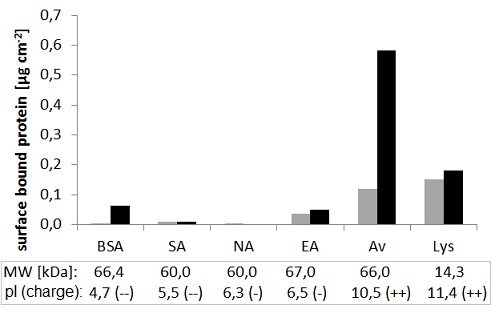
Figure 9: Adsorption of different proteins on MIP (black) and NIP (grey).
Molecular weight (MW) and isoelectric point (pI), as well as the charge at the applied pH (7.4) are shown.
Expected impact and further research
We have developed a novel method for molecular imprinting based on nanosphere lithography, which yielded an outstanding imprinting factor and a remarkable selectivity against analogues of the target protein. This work has contributed to the scientific program titled Bio(chemical sensing with functionalized nanostructures (TÁMOP-4.2.1/B-09/1/KMR-2010-0002 NNA-P4-T1). Our results have been featured on the front page of Advanced Functional Materials (IF2012=9,765) [B3], and were presented at various Hungarian and international conferences [B4-9].
An advantage of nanosphere-lithography based molecular imprinting is the ability to tune the binding site density in the formed polymer film by adjusting the protein-coverage of the template beads. We are currently developing a modified drop-casting method to achieve a compact layer from the protein-modified beads; this is expected to further improve the analytical perfomance of our MIPs. Our aims include the use of magnetic beads which would enable template recovery, thus making the procedure more economical.
Publications, references, links
Publications
[B1] Horváth, V., Lorántfy, B.; Tóth, B.; Bognár, J.; László, K.; Horvai, G., J. Sep. Sci., 2009, 32 (19) 3347. IF=2,551
[B2] Bognár, J.; Szűcs, J.; Dorkó, Z.; Horváth, V.; Gyurcsányi, R.E., Adv. Funct. Mater., 2013, 23 (37) 4703. IF=9,765 (2012)
[B3] Bognár, J.; Szűcs, J.; Dorkó, Z.; Horváth, V.; Gyurcsányi, R.E., Adv. Funct. Mater., 2013, 23 (37) 4641.
Conference presentations
[B4] Bognár, J.; Horváth, V.; Gyurcsányi, R.E.: Towards surface-imprinted nanostructures for selective protein recognition. Graduate Student Symposium on Molecular Imprinting, 2011, London, UK (poster)
[B5] Bognár, J.; Lautner, G.; Syritski, V.; Horváth, V.; Gyurcsányi, R.E.: Surface-imprinted nanostructures for selective protein recognition. Mátrafüred 2011 International Conference on Electrochemical Sensors, 2011, Dobogókő, Hungary (poster)
[B6] Bognár, J.; Szűcs, J.; Gyurcsányi, R.E.; Horváth, V.: Towards surface-imprinted nanostructures for selective protein recognition. 7th International Conference on Molecularly Imprinted Polymers – Science and Technology, 2012, Paris, France (poster)
[B7] Bognár, J.; Szűcs, J.; Dorkó, Z.; Horváth, V.; Gyurcsányi, R.E.: Nanosphere lithography as a versatile method to generate surface-imprinted polymer films for selective protein recognition. Graduate Student Symposium on Molecular Imprinting, 2013, Belfast, UK (oral presentation)
[B8] Bognár, J.; Szűcs, J.; Dorkó, Z.; Horváth, V.; Gyurcsányi, R.E.: Szelektív fehérje-felismerés nanoszféra-litográfián alapuló molekuláris lenyomatképzéssel. A szenzorkutatás újabb eredményei Workshop V., 2013, Pécs, Hungary (oral presentation)
[B9] Bognár, J.; Szűcs, J.; Dorkó, Z.; Horváth, V.; Gyurcsányi, R.E.: Selective protein recognition with surface molecularly imprinted polymer films prepared by nanosphere lithography. Mátrafüred 2014 International Conference on Electrochemical Sensors, 2014, Visegrád, Hungary (poster)
Links
Nanosphere (or colloidal) lithography
References
[1] Ferrer, I.; Lanza, F.; Tolokán, A.; Horváth, V.; Sellergren, B.; Horvai, G.; Barceló, D., Anal. Chem. 2000, 72 (16) 3934.
[2] Pap, T.; Horváth, V.; Tolokán, A.; Horvai, G.; Sellergren, B., J. Chrom. A, 2002, 973 (1-2) 1.
[3] Bereczki, A.; Tolokán, A.; Horvai, G.; Horváth, V.; Lanza, F.; Hall, A.J.; Sellergren, B., J. Chrom. A, 2001, 930 (1-2) 31.
[4] Pap, T.; Horvai, G., J. Chrom. A, 2004, 1034 (1-2) 99.
[5] Pap, T.; Horvai, G., J. Chrom. B, 2004, 804 (1) 167.
[6] Tóth, B.; László, K., Horvai, G., J. Chrom. A, 2005, 1100 (1) 60.
[7] Tóth, B.; Pap, T.; Horváth, V.; Horvai, G., J. Chrom. A, 2006, 1119 (1-2) 29.
[8] Tóth, B.; Pap, T.; Horváth, V.; Horvai, G., Anal. Chim. Acta, 2007, 591 (1) 17.
[9] Tóth, B.; Pap, T.; Horváth, V.; Horvai, G., Magyar Kém. Foly., 2005, 111 (3) 110.
[10] Lanza, F.; Hall, A.J.; Sellergren, B.; Bereczki, A.; Horvai, G.;Bayoudh, S.; Cormack, P.A.G.; Sherrington, D.C., Anal. Chim. Acta, 2001, 435 (1) 91.
[11] Ceolin, G.; Navarro-Villoslada, F.; Moreno-Bondi, M.C.; Horvai, G.; Horváth, V., J. Comb. Chem., 2009, 11 (4) 645.
[12] Renkecz, T.; László, K.; Horváth, V., J. Mol. Recogn., 2012, 25 (6) 320.
[13] Ceolin, G.; Orbán, Á.; Kocsis, V.; Gyurcsányi, R.E.; Kézsmárki, I.; Horváth, V., J. Mater. Sci., 2013, 48 (15) 5209.
[14] Menaker, A.; Syritski, V.; Reut, J.; Öpik, A.; Horváth, V.; Gyurcsányi, R.E., Adv. Mater., 2009, 21 (22) 2271.
[15] Lautner, G.; Kaev, J.; Reut, J.; Öpik, A.; Rappich, J.; Syritski, V.; Gyurcsányi, R.E., Adv. Funct. Mater., 2011, 21 (3) 591.
[16] Vezenov, D.V.; Noy, A.; Rozsnyai, L.F.; Lieber, C.M., J. Am. Chem. Soc., 1997, 119 (8) 2006.
[17] http://www.personal.psu.edu/acs5112/ART101/art101ass5.html