|
|
BMe Research Grant |
|

Szigetvári Áron
BMe Research Grant - 2022
IInd Prize
![]()
George A. Olah Doctoral School of Chemistry and Chemical Technology
Spectroscopic Research Department, Gedeon Richter Plc.
Supervisor: Dr. Szántay Csaba
Efficiency-increased NMR experiments in pharmaceutical industrial applications of structure elucidation
Introducing the research area
Although two-dimensional (2D) NMR spectroscopy provides an exceptionally rich set of information in the elucidation of the chemical structure of small molecules, harvesting its full capabilities in the pharmaceutical industry was rather cumbersome until the recent past, when the application of an acquisition accelerating method, namely nonuniform sampling (NUS), has become routine. Since we are both required, and also committed to providing state-of-the-art structural research in a pharmaceutical R&D and quality control setting, lately we have been actively exploring the advantages of NUS. As a result of our research in this area, we have demonstrated for the first time that NUS, combined with a known spectrum simplifying technique, effectively supports spectroscopists in solving the most difficult structural questions with high confidence.
Brief introduction of the research place
The main field of the Spectroscopic Research Department, Gedeon Richter Plc., is small-molecule structure elucidation, mainly based on the concerted use of NMR spectroscopy and mass spectrometry. Full-time spectroscopists constantly look for the best available methods in the scientific literature and adapt them to the pharmaceutical industry. We strive to come up with innovative solutions [1] to real-world problems in the field of structural research.
History and context of the research
The relevance of NMR spectroscopy in the pharmaceutical industry can probably be best appreciated from the point of view of quality control. Since the quality of pharmaceuticals is strictly regulated by the authorities, the consistent production of high-purity (typically of >99.5 weight%) drug substances and drug formulations is an absolute necessity. In addition, drug companies are obliged to identify the structure of any trace impurity that can be found in an amount of >0.10% of the drug substance. Fulfilling such a task can be rather challenging, and it requires the collaboration of numerous methods and fields of scientific expertise. First, analytical chemists demonstrate the presence of a trace impurity which is then isolated by preparative chromatographers, yielding a mixture in which the amount of impurity is enhanced by typically two orders of magnitude. Afterward, the mixture is submitted for structural analysis. Finally, synthetic chemists synthesize the substance with the elucidated structure in larger quantities to provide analytical chemists with a suitable analytical standard for quantitative analysis regulated according to current Good Manufacturing Practice.
In the pharmaceutical industry, the stakes of structural analysis are rather high, as an erroneous structural proposal can generate an immense financial and reputational loss. A faulty structural proposal for a trace impurity may cause the short-term (e.g., a few months) efforts of a smaller group of synthetic chemists to go to waste. In other research projects, for example, drug discovery, an erroneous chemical structure may corrupt the models used for following structure-activity relationships, resulting in temporary dead-ends in the search for a new chemical entity. Therefore, effective, and accurate determination of any molecular structure is critical in the whole pharmaceutical ecosystem.
The research goals, open questions
Due to clientele pressure on the analysis time of the samples (which is a characteristic of an industrial R&D setting), spectroscopists are constantly facing the dilemma of being quicker or more thorough. It is essential to manage the instrumental capacity well, so in case of large sample throughput, only some basic NMR measurements can be performed. On the other hand, we must avoid any structural misassignments, but the way to higher confidence in the structural proposals is the acquisition of larger sets of spectral data. The main question of our research is to what extent one can deal with these contradictory demands by using state-of-the-art NMR techniques.
Another stress factor in structure determination relates to the fuzziness in the interpretation of some spectral data. For example, one can detect some kind of connection between a hydrogen atom and a carbon atom in the HMBC spectrum (vide infra), but there are no reliable methods for determining that the atoms are two bonds or three bonds away from each other. Such decisions demand additional measurements that take up instrumental capacities. Alternatively, one can rely on the chemical context (starting materials, reagents, and solvents used for the preparation of the sample) of the structural problem during the interpretation of the NMR spectra. However, it is relatively easy (especially under time pressure) to fall into traps of overusing chemical context-related structural premises. In such cases, one may dismiss some ambiguities during data interpretation and support a chemically rational, but in fact wrong structural proposal.[2]
Our research aimed at incorporating the use of nonuniformly sampled 2D NMR spectra in pharmaceutical structural analysis. We persist with our dual commitment to being quick and thorough; therefore, we explored the ultimate capabilities of NUS in accelerating NMR measurements in the context of pharmaceutical applications.
Methods
Typically, six orthogonal NMR techniques are applied in the structure determination of pharma-related samples (Figure 1). These are 1H NMR spectroscopy, 13C NMR spectroscopy, and four kinds of 2D NMR techniques aiming at detecting various types of connections between nuclei. The latter ones are COSY (detection of 1H-1H connections via, most often three, chemical bonds), NOESY (detection of 1H-1H spatial proximity), HSQC (direct connection of 1H and 13C nuclei), and HMBC (1H-13C connections via multiple chemical bonds). We note that NMR spectroscopists deduce the presence of other atoms (e.g., oxygen) indirectly by using 1H NMR and 13C NMR chemical shifts, which is ambiguous to some extent. Therefore, we need to collaborate with mass spectroscopists who gather information about the molecular weight and the elemental composition of a compound.
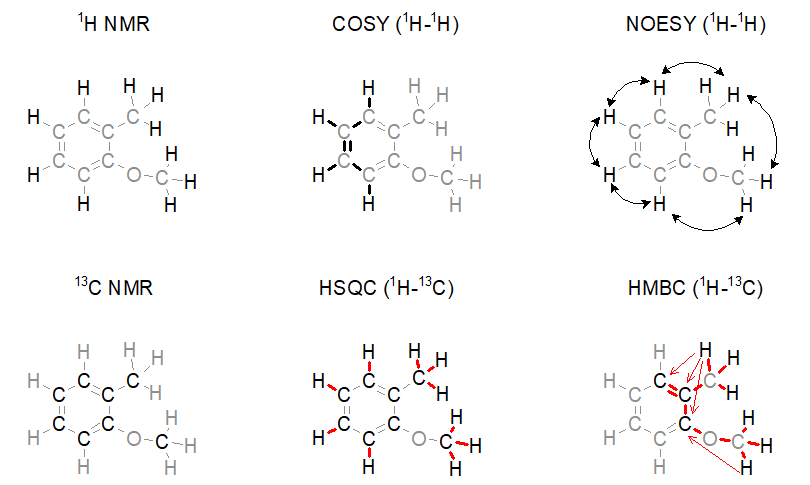
Figure 1. Widely used NMR spectroscopic methods for the determination of the structure of small molecules.
The capabilities of the methods shown in Figure 1 depend on two factors. On the one hand, one cannot quickly acquire all kinds of spectra with an adequate signal-to-noise ratio when only a limited amount (ca. 1–5 mg) of sample is available. For example, carbon NMR spectroscopy is an insensitive experiment (Figure 2a.) because 12C is magnetically inactive and the only measurable isotope is 13C which is present only in 1%. On the other hand, in the case of complex samples, overlapping of the peaks may introduce ambiguities in the interpretation of spectral data. The resolution of the 1H NMR spectrum can be improved by using a stronger NMR magnet. However, the resolution in 2D NMR spectra can only be increased by measuring additional (sometimes, several thousands of) data points (Figure 2b.); the problem persists independently of sample amount and hardware capabilities. Therefore, 2D NMR measurements are often slow (several seconds per data point); the analysis of complex structures requires lengthy (hours of) data acquisition. In routine applications, the acquisition of the four 2D NMR spectra (COSY, NOESY, HSQC, and HMBC) lasts for one to two hours on average.
The main feature of NUS is that a relatively large portion of data points (intensity versus time samples) can be omitted from an NMR measurement; the position of the peaks (a frequency-like quantity) can still be reconstructed from the remaining subset of data points (Figure 2c.). It is analogous to the determination of the frequency of a sine wave: if we are sure that the signal is a sinusoid and we know the range of the possible frequencies, the accurate frequency of the wave can be calculated from a few intensity-time points. Undersampled versions of a more complex signal composed of multiple frequency components (corresponding to multiple peaks in a spectrum) can also be an adequate representation of the signal provided that the number of sampled data points is several times larger than the number of expected frequency components.[3]
Consequently, one can acquire a high-resolution 2D NMR spectrum (corresponding to thousands of data points) by randomly omitting the sampling of a portion (up to more than 90%) of the scheduled data points and by using spectrum reconstruction software for guessing the missing data points. Spectrometer manufacturers have included the appropriate algorithms for the reconstruction of NUS 2D NMR spectra in their standard software packages in the last decade (earlier, the use of NUS was limited to structural biology NMR applications).
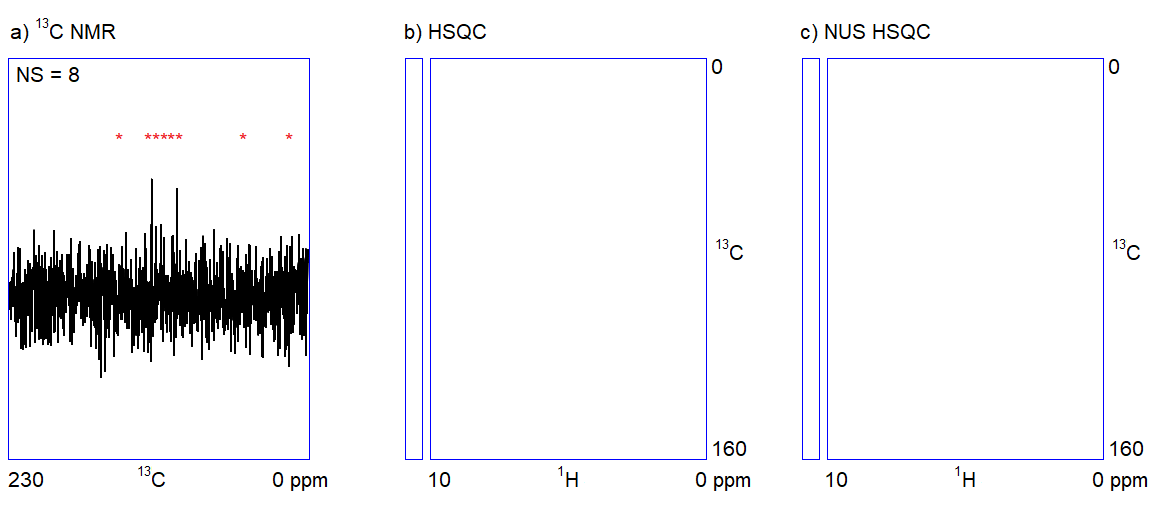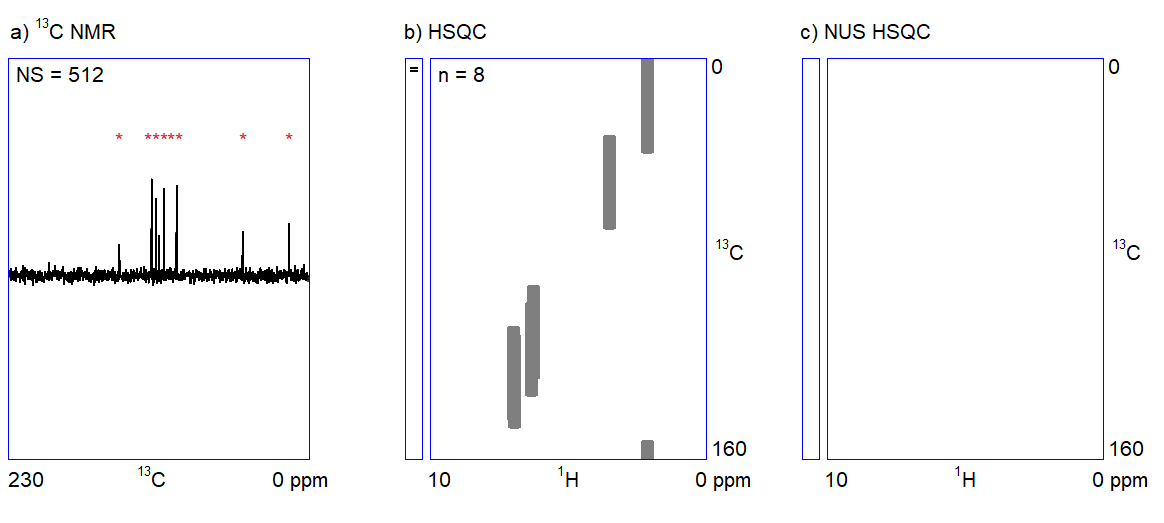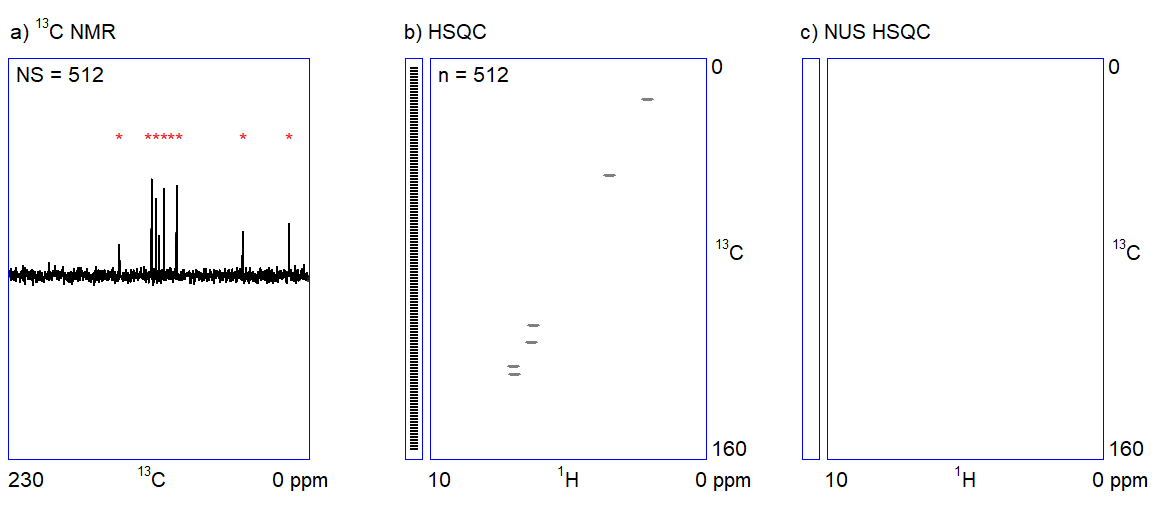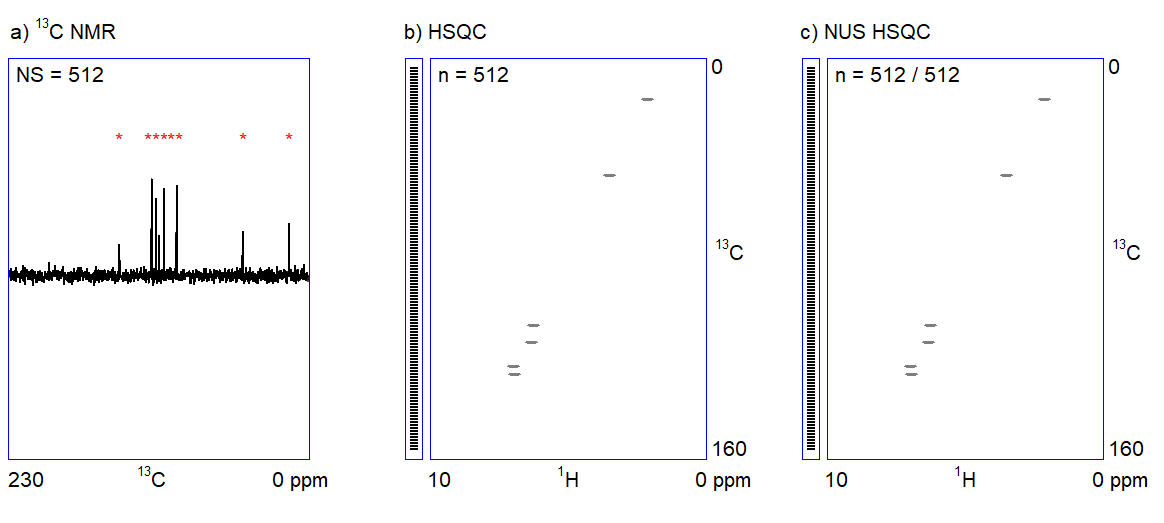
Figure 2. a) Snapshots of the 13C NMR measurement of a dilute sample; NS = number of scans; b) Traditionally sampled HSQC spectrum of the sample; n = number of data points (see the narrow bar to the left of the spectrum); c) Nonuniformly sampled HSQC spectrum of the same compound; n = number of selected data points (the distribution of the selected data points are shown in the narrow bar to the left of the spectrum).
During our research project, we optimized the NUS parameters (the acquisition of how many data points should be planned and from that set how many can be skipped) for some applications of NMR spectroscopy in a pharmaceutical industrial context. We observed that the fine structure of spectral peaks limits the application of NUS in extremely high-resolution spectra. To overcome the limitations, we have combined NUS with a known peak simplifying technique [4] for the first time.
Results
In a collaboration with a synthetic chemistry group dealing with the synthesis of vinblastine-like analogues, our partners isolated an unexpected and odd by-product, which turned out to be a vindoline trimer consisting of 188 atoms (C74H90N6O18). The structure elucidation of the by-product was particularly difficult because the NMR signals were overlapping due to the three subunits; there were many peaks of ambiguous meaning in the traditionally sampled NMR spectra. We used NUS for resolving overlapping peaks and performed the measurements on an 800 MHz NMR spectrometer equipped with a cryogenically cooled probe. We conducted the HSQC experiment (see Methods part) by only sampling 256 data points out of 8192 points (by skipping the acquisition of 97% of the data points), reducing the acquisition time from 6 hours to 11 minutes. The HMBC spectrum was measured by sampling 512 points out of 8192 points (by skipping the acquisition of 94% of the data points), reducing the acquisition time from 12 hours to 47 minutes. We deduced the structure of the vindoline trimer from a thousand pieces of information in the resulting NMR spectra (a part of that process is shown in Figure 3).[S2]
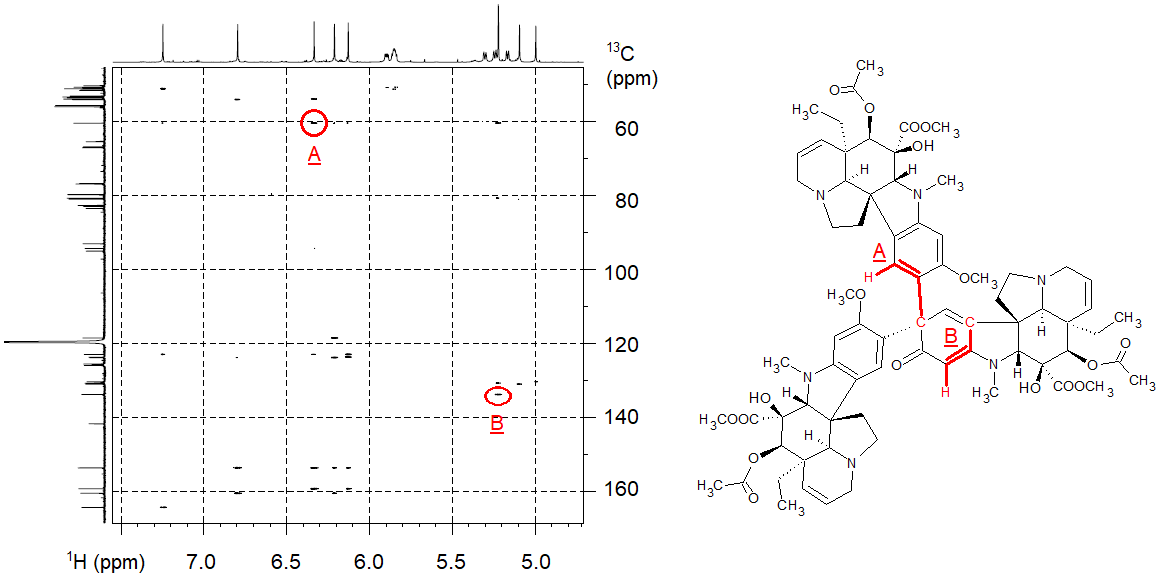
Figure 3. High-resolution HMBC spectrum acquired with the aid of NUS (the axes represent 1H and 13C NMR chemical shifts); the interpretation of two selected HMBC peaks is shown in the structure.
We have set up a measurement protocol by maximizing the utility of NUS in practical routine spectroscopic applications (providing analytical support for chemical synthesis). For relatively concentrated samples, the six NMR spectra shown in Figure 1 can be recorded within 21 minutes instead of an hour or two hours characteristic of the pre-NUS era.[S3] For dilute samples, we recalculate the appropriate NUS acquisition parameters; we made a prototype script that automates the calculations and starts the experiments with only a little user intervention. We managed to increase the speed of our structure elucidation service and decrease the amount of stress on the spectroscopists while maintaining high confidence in the proposed structures.
The most significant novelty of our work is the idea of combining NUS with a known technique called “constant time” (CT) that eliminates some features of the fine structure of the peaks in 2D NMR spectra. Our method performs well for spectra consisting of highly congested regions, such as the HMBC spectrum of a tetrakis(propargyl ether) compound. The eight carbon atoms involved in triple bonds gave signals within a very narrow chemical shift range (1 ppm wide range of a ca. 200 ppm wide 13C NMR chemical shift range). When we attempted to acquire a high-resolution HMBC spectrum of the sample by using NUS, the resulting spectrum was unintelligible due to the ill-reconstructed fine structure of the HMBC peaks (Figure 4a), so we were not able to assign the 13C NMR resonances to the carbon atoms in the structure. However, our idea of combining NUS with CT resulted in a significantly cleaner HMBC spectrum (Figure 4b); therefore, we were able to prove the correct connection of the atoms in the four propargyl groups (denoted as P1…P4).[S3]
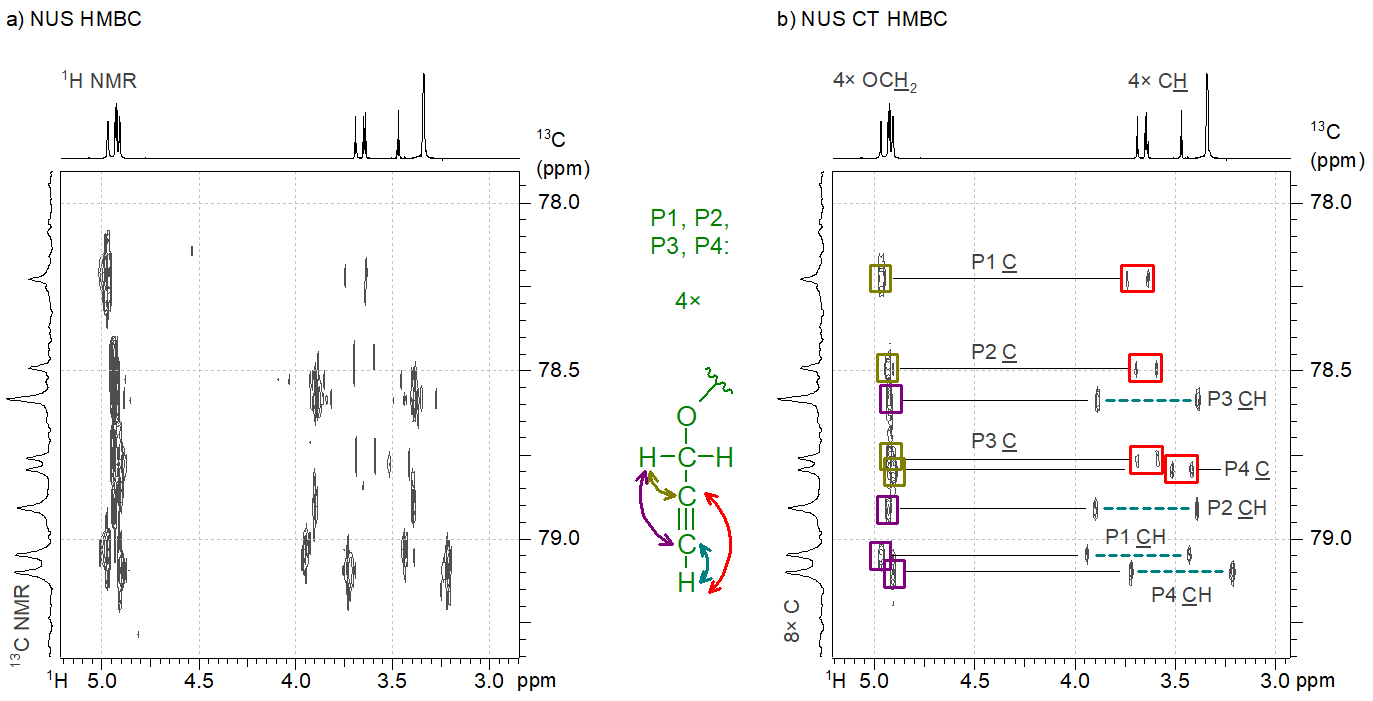
Figure 4. a) The nonuniformly sampled HMBC spectrum of a tetrakis(propargyl ether compound); b) The constant time NUS HMBC spectrum of the same compound.
We successfully applied our novel methodology during trace impurity analysis of a starting material used for drug production. By using the CT NUS HMBC and the NUS HSQC spectra recorded from the starting material, we determined the structure of an impurity that was present in the sample at a 0.14% level (Figure 5). By using traditional spectrum acquisition methods, we could not reveal the presence of a constitutional isomer as its spectral peaks were covered by the peaks belonging to the main compound. Additional examples of the application of NUS and CT in the pharmaceutical industry can be found in our publication.[S3]
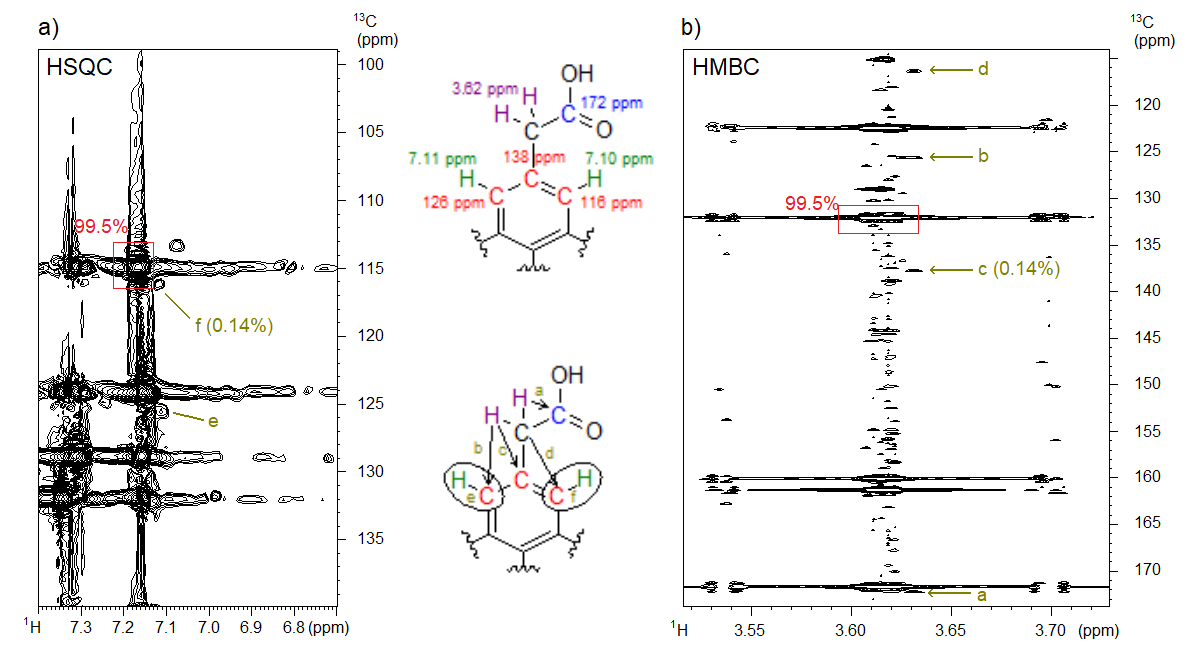
Figure 5. a) The NUS HSQC and b) the NUS CT HMBC spectrum of the starting material containing 0.14% of the denoted structural isomer impurity at 800 MHz. The 1H NMR and 13C NMR chemical shifts (corresponding to the position of the peaks in the spectra) of selected atoms are shown in the upper structure; in the lower structure, the interpretation of some selected spectral peaks (a–f) is explained.
Expected impact and further research
After the publication of our results in the scientific literature, we further developed our script that automates the calculation of the NUS acquisition parameters and launches the NMR experiments. Our automation script has saved us several hundred working hours. We are committed to monitoring the literature in order to introduce state-of-the-art NMR methodologies at Gedeon Richter. Our new idea of combining nonuniform sampling with the constant time technique can facilitate the elucidation of the most complex structures in other NMR laboratories. So far, our paper has received one citation from an independent research group.
Publications, references, links
List of corresponding own publications.
[S1] Szigetvári Á., Szántay Cs. Szerkezetkutatás NMR-spektroszkópiával a gyógyszeriparban: hatékonyságnövelés nem egyenletes mintavételezés segítségével. Magy. Kém. Foly. 2020, 126, 135–146.
[S2] Á. Szigetvári, A. Keglevich, P. Keglevich, M. Dékány, L. Hazai, C. Szántay, Jr. A mainly NMR-based structure elucidation of a surprising vindoline trimer with the aid of non-uniform sampled 1H-13C HSQC and HMBC spectra. Struct. Chem. 2021, 30, 795–804. (IF: 1.887)
[S3] Á. Szigetvári, C. Szántay, Jr. On the utility of non-uniformly sampled two-dimensional NMR spectra in the pharmaceutical industry. Magn. Reson. Chem. 2021, 59, 264–286. (IF: 2.447)
[S4] Szigetvári Á., Keglevich A., Pápai R., Dékány M., Béni Z., Keglevich P., Hazai L., Szántay Cs. A vinka alkaloidok szerkezetkutatása az NMR spektroszkópia szemszögéből – két érdekes esettanulmány. Alkaloid- és Flavonoidkémiai munkabizottság, Mátrafüred, 2016.
[S5] Szigetvári Á., Keglevich A., Pápai R., Dékány M., Keglevich P., Hazai L., Béni Z., Szántay Cs. A vinka alkaloidok szerkezetkutatása az NMR spektroszkópia szemszögéből - két érdekes esettanulmány. Fiatal Analitikusok Előadóülése, Budapest, 2016.
[S6] Szigetvári Á. A maradékos osztás (spectral aliasing) alkalmazása egy meglepő vindolin trimer szerkezetmeghatározására. A Magyar NMR Munkabizottság ülése, Debrecen, 2016.
[S7] Á. Szigetvári, A. Keglevich, M. Dékány, P. Keglevich, Z. Béni, L. Hazai, C. Szántay, Jr. NMR-based structure elucidation of a surprising trimeric vindoline product with the aid of heteronuclear spectral aliasing methods. Magnetic Moments in Central Europe, Budapest, 2017.
[S8] Szigetvári Á. Az NMR spektroszkópia alkalmazásának sokszínűsége a gyógyszeripari kutatásban. I. Fiatal Kémikusok Fóruma (FKF) Szimpózium, Debrecen, 2019.
[S9] Szigetvári Á., Szántay Cs. Szerkezetkutatás NMR-spektroszkópiával a gyógyszeriparban: hatékonyságnövelés nemegyenletes mintavételezés segítségével. Fiatal Analitikusok Előadóülése, online, 2020.
[S10] Á. Szigetvári, C. Szántay, Jr. On the utility of nonuniformly sampled two-dimensional NMR spectra in the pharmaceutical industry. Magnetic Moments in Central Europe, Primošten, Croatia, 2022.
Table of links.
Two-dimensional (2D) NMR spectroscopy
Authority regulations of pharmaceuticals
An erroneous structural proposal (the consequences of ~)
Structure-activity relationships
Traps, mental traps (in science)
800 MHz NMR spectrometer (video)
List of references.
[1] Z. Szeleczky, Z. Szakács, É. Bozó, F. Baska, K. Vukics, S. Lévai, K. Temesvári, E. Vass, Z. Béni, B. Krámos, I. Magdó, C. Szántay Jr., J. Kóti, K. Domány-Kovács, I. Greiner, I. Bata. Synthesis and Characterization of New V1A Antagonist Compounds: The Separation of Four Atropisomeric Stereoisomers. J. Med. Chem. 2021, 64, 10445–10468.
[2] C. Szantay, Jr. (szerk.) Anthropic Awareness – The Human Aspects of Scientific Thinking in NMR Spectroscopy and Mass Spectrometry. Elsevier, 2015.
[3] A. Shchukina, P. Kasprzak, R. Dass, M. Nowakowski, K. Kazimierczuk. Pitfalls in compressed sensing reconstruction and how to avoid them. J. Biomol. NMR, 2017, 68, 79–98.
[4] K. Furihata, H. Seto. Constant time HMBC (CT-HMBC), a new HMBC technique useful for improving the separation of cross peaks. Tetrahedron Lett. 1998, 39, 7337–7340.